Background
Subdural empyema (SDE) represents a rare but life-threatening intracranial infection in the pediatric and adolescent population. Epidemiologic patterns are influenced by age, sex, underlying risk factors, and geographic differences. SDE is characterized by purulent accumulation in the subdural space, usually secondary to bacterial spread from meningitis, sinusitis, or otitis media.1,2 Clinical manifestations of subdural empyema are diverse, often including fever, mental status changes, focal neurological deficits, seizures, and features of increased intracranial pressure. Prompt neurosurgical intervention is critical due to the risk of rapid deterioration and serious complications.3 Microbiologically, subdural empyema is typically polymicrobial, with Streptococcus anginosus group, staphylococci, anaerobes and, less commonly, aerobic gram-negative bacilli implicated.4 In rare cases, unusual pathogens such as Streptococcus constellatus have been isolated, even in immunocompetent children.5 This report presents a rare case of hemophagocytic lymphohistiocytosis associated with subdural empyema secondary to Streptococcus constellatus.
Case presentation
A 16-year-old male presented to the hospital complaining of right eye pain, holocranial headache, fever, and vomiting that started eight days prior and worsened significantly in the last three days. He denied any allergies, smoking, taking medications at home, or using illicit drugs. There was no history of recent travel or trauma. He reported symptoms of chronic sinus problems, including nasal congestion, frontal headaches, low-grade fever, and frequent nocturnal cough. There was also no family history of neoplasms or spinal problems. On physical examination, he achieved a Glasgow Coma Scale (GCS) score of 15. His pupils were reactive to light and isochoric, and he showed no focal deficits; however, he had periorbital edema on the right side. Due to suspicion of periorbital cellulitis, empirical antibiotic therapy with oxacillin and ceftriaxone was initiated.
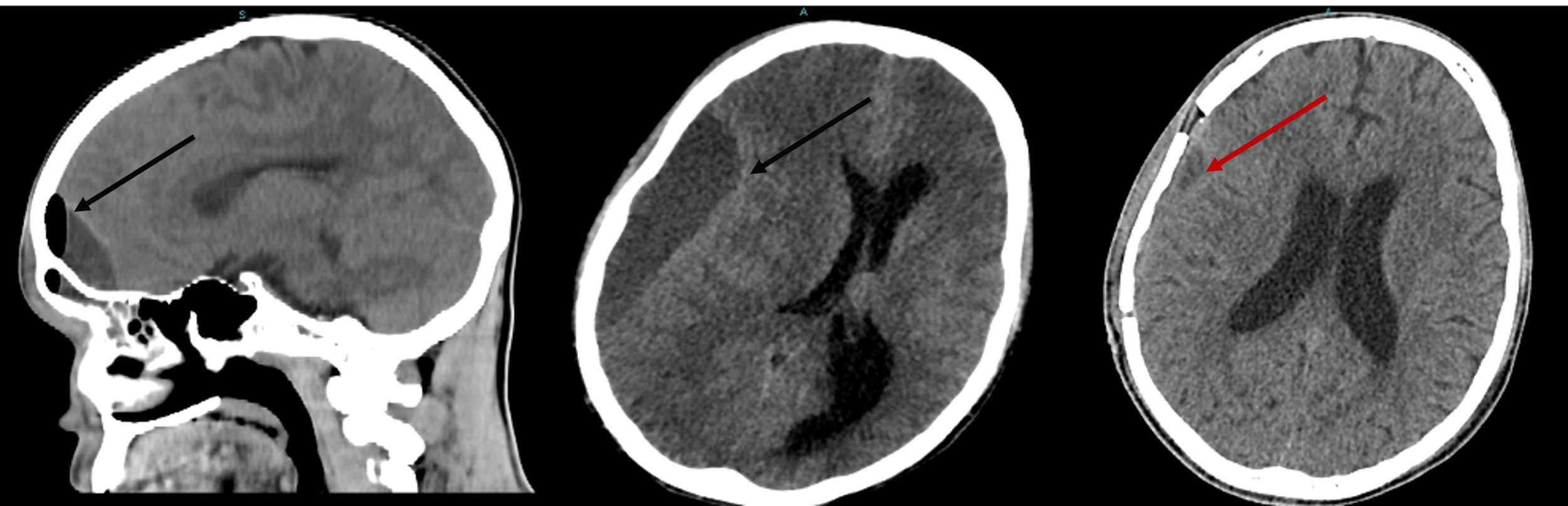
A non-contrast computed tomography scan (CT) of the brain revealed a pansinusopathy and a right frontal collection 12.0 cm3 in size. These findings were suggestive of extradural empyema. Although contrast-enhanced magnetic resonance imaging (MRI) is generally considered superior for diagnosis and surgical planning, MRI was not performed in this case due to limited availability and the urgent need for immediate treatment. The patient underwent urgent surgical drainage of extradural empyema by right supraorbital mini craniotomy. A wound culture obtained during surgery revealed multidrug-resistant Streptococcus constellatus; the antibiotic regimen was subsequently changed to cefepime and vancomycin. On day 15, the patient developed decreased consciousness with a GCS of 6, prompting a CT brain that revealed a right-sided subdural empyema requiring surgical drainage. The procedure was uneventful, and a follow-up CT performed 10 days later showed progressive clinical and neurological improvement during hospitalization. (Figure 1).

Amid ongoing antibiotic therapy in the hospital, the patient developed fever, pallor, and jaundice. Physical examination revealed the spleen palpable 3–4 cm below the costal margin, without evidence of lymph node enlargement. Laboratory workup demonstrated cytopenia and hepatic dysfunction, evidenced by elevated transaminases (AST 500.6 IU/L; ALT 1682 IU/L). Over the course of his hospital stay, his WBC count reached a low of 1,240/µL. Additionally, the abdominal ultrasound revealed homogeneous splenomegaly, measuring 15.2 x 7 cm. There was no clinical or laboratory evidence of a new systemic infection. There was no evidence of arthritis, neurological deterioration, or sepsis. The brain CT scan showed no new intracranial collections.
His serum ferritin was elevated at 647.2 (ng/ml) (reference value up to 200 ng/ml for men according to the World Health Organization). However, triglycerides, syphilis tests, and serological tests for hepatitis B and C and HIV were within normal limits. A panel for viral antibodies was also performed, which included Epstein-Barr virus (EBV), cytomegalovirus (CMV), and herpes simplex virus (HSV). All of these viruses were undetectable in the serum, except for HSV, where IgG antibodies were detected.
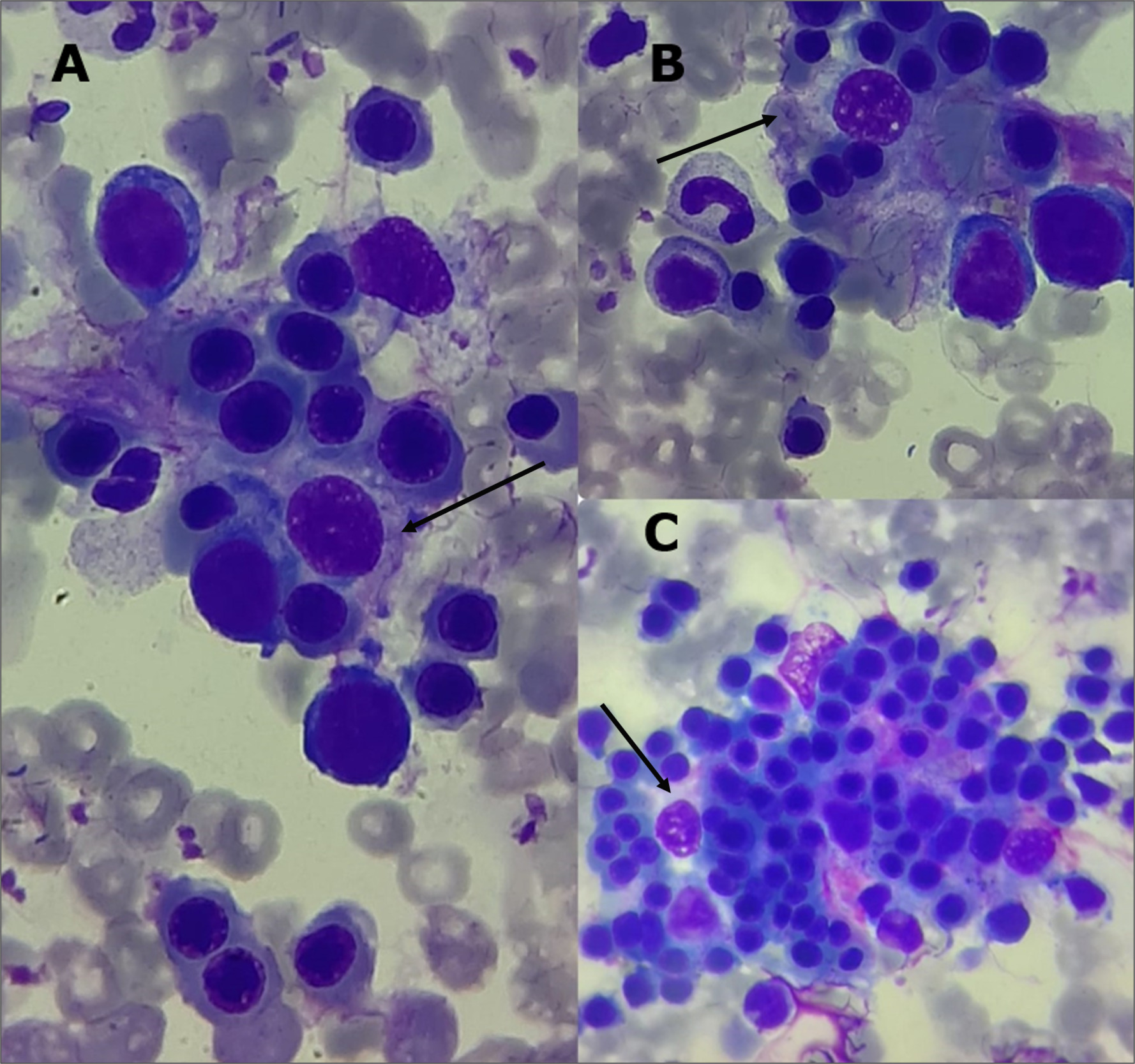
Given concern for hemophagocytic lymphohistiocytosis (HLH), a bone marrow aspiration was performed, revealing a slight hypercellularity, primarily affecting the erythroid and megakaryocytic lineages. A marked increase in macrophage activity was observed, accompanied by evidence of hemophagocytosis (Figure 2). A spinal tap was also performed. Cerebrospinal fluid (CSF) was slightly turbid, with glucose 61 mg/dL (normal: 45–80 mg/dL), protein 24.9 mg/dL (normal: 15–45 mg/dL), 1,900 RBCs/mm³, and 7 WBCs/mm³ (normal: 0–5; 29% neutrophils, 71% lymphocytes). CSF cultures were negative. Blood cultures also showed no microbial growth.

The differential workup included severe sepsis, acute leukemia, macrophage activation syndrome due to autoimmune disease, and systemic viral infections (HIV, HBV, HCV, CMV, EBV). These were ruled out by negative blood cultures, a comprehensive negative viral serology panel, bone marrow aspirate excluding acute leukemia, and the absence of autoimmune or rheumatologic features. In view of the clinical presentation consistent with sHLH and disease severity, treatment with dexamethasone (10 mg/m²) was started in combination with antibiotics. The patient was stable, without signs of respiratory compromise or neurological involvement. Corticosteroid therapy led to significant clinical and laboratory improvement, with normalization of abnormal parameters.
Discussion
HLH has a primary (familial autosomal recessive or genetic) or a secondary form. In primary HLH, genetic alterations affecting lymphocyte cytotoxicity are involved, and some of these genetic changes are also responsible for certain albinism syndromes.6–8 The case described refers to a secondary condition that occurs in adults and may be triggered by an infection. The early symptoms of sHLH are often non-specific and can overlap with other inflammatory or hematologic disorders. Initial investigations must focus on establishing a prompt diagnosis of sHLH while identifying treatable triggers or mimicking conditions.7,9–11
HLH typically presents with recurrent fever, cytopenias, liver dysfunction, and a sepsis-like syndrome that may quickly progress to multiorgan failure.11,12 Fever is often above 38.5 degrees Celsius and does not respond to antibiotic therapy. The disease is characterized by hepatosplenomegaly, coagulopathy, neurological disturbances, and elevated biomarkers, such as ferritin and soluble interleukin 2 receptor.11–14 In addition, non-specific clinical manifestations such as edema, skin rashes, and gastrointestinal symptoms may also occur. Involvement of the lungs, kidneys, or heart is possible in severe cases. Neurological symptoms are more common in pediatric cases and include seizures, meningismus, peripheral neuropathy, cranial nerve involvement, ataxia, dysarthria, lethargy, encephalopathy, and even isolated first presentation.11
Streptococcus constellatus, a member of the Streptococcus anginosus group, is part of the normal oral and upper respiratory tract flora, but it is known for its propensity to cause pyogenic infections, including subdural empyema.15 The pathogenesis of SDE is frequently linked to the spread of infections such as sinusitis, otitis media, or odontogenic infections. Streptococcus constellatus and other polymicrobial oral organisms may act as causative agents due to their ability to invade deep tissues and form abscesses, including within the central nervous system in young individuals.15 In our case, increased social vulnerability may have contributed to this complication, as limited access to basic sanitation, primary healthcare, health education, adequate nutrition, and food security are key determinants of infection risk and outcomes. To our knowledge, hemophagocytic lymphohistiocytosis associated with Streptococcus constellatus infection has not been reported in the literature, in contrast to its association with Streptococcus intermedius infections (another member of the Streptococcus anginosus group), although this is also rare.16 However, the pathophysiological mechanism is the same, as this pathogen (S. constellatus) can behave pathogenically by invading tissues and infecting the central nervous system.
The Histiocyte Society has established consensus criteria for the HLH-94 and HLH-2004 clinical trials, which include the genetic basis of familial HLH.17 Although these criteria have consolidated over the past decade as the basis for defining HLH, their sensitivity and specificity still need to be discovered, and an updated international consensus based on evidence is needed. At least five of the eight criteria must be met for the diagnosis to be made.11 In special situations, therapy directed at HLH can be initiated with modified criteria that require three of the four major clinical findings (fever, splenomegaly, cytopenias, hepatitis) and an abnormality in one of the four immunological markers (hemophagocytosis, elevated ferritin, hypofibrinogenemia, or decreased NK cell function).1,5,12 It is essential to note that meeting the criteria for HLH does not rule out the possibility of an underlying infection (primarily Epstein-Barr virus) or associated malignancy.5
In our case, additional immunologic tests such as soluble IL-2 receptor, NK cell function, and fibrinogen levels could not be performed because they were not available at our institution at the time of diagnosis. Nevertheless, the patient had fever, splenomegaly, cytopenias, hepatitis, hyperferritinemia, and hemophagocytosis in the bone marrow, thus fulfilling the modified criteria for HLH. These modified criteria have been proposed to enhance sensitivity in urgent clinical scenarios, allowing treatment to be initiated even when a full laboratory panel is not available.11,13 Given the high risk of rapid progression and fatal outcome, treatment was initiated without delay, which likely contributed to the favorable response.
In addition, other viruses such as HIV, parvovirus, herpes simplex virus (HSV) (especially in neonates), varicella zoster virus (VZV), measles, human herpes virus (HHV)-6, HHV-8, H1N1 influenza virus, parechovirus, parvovirus, and dengue virus have also been associated with HLH.18,19 Recently, an association between HLH and COVID-19 infection has been documented in the literature.20 Less commonly, HLH may be associated with bacterial infections, including those caused by gram-negative bacteria and Mycobacterium tuberculosis, as well as fungal pathogens such as Histoplasma capsulatum. In addition, neglected diseases such as malaria and leishmaniasis are rarely associated with HLH.19 In the present case, as mentioned, the patient developed a bacterial infection (subdural empyema) as a trigger, being a condition with rare cases in the literature.
Treatment for HLH should be initiated as soon as the syndrome is recognized after other similar conditions have been ruled out. Patient survival has improved significantly with the HLH-94 protocol, with a long-term survival rate exceeding 50%.7 The essential treatment consists of etoposide and steroids, with dexamethasone as a first option in some cases.8,10 The patient responded positively to the isolated use of dexamethasone in conjunction with infection control.4 Although cyclosporine has shown benefit in studies, the HLH-94 protocol remains the recommended standard of care.9,13
Conclusion
The report presents a compelling case of a disease characterized by substantial morbidity and mortality, alongside a rare etiology (S. constellatus) in an immunocompetent young patient. Recognizing the presence of this condition and swiftly implementing appropriate therapeutic interventions, even in scenarios where diagnostic capacities are suboptimal, emerges as a pivotal factor in achieving favorable treatment outcomes. This case report is poised to offer invaluable insights to healthcare professionals engaged in patient care and to catalyze further research exploration into this intricate and challenging medical condition.
Disclosures/Conflicts of Interest
The authors declare that they have no conflicts of interest
Corresponding author:
Jandir Mendonça Nicacio M.D; MSc; Ph.D,
Department of Medicine,
Universidade Federal do Vale do São Francisco, Brazil
Email: jandir.nicacio@univasf.edu.br