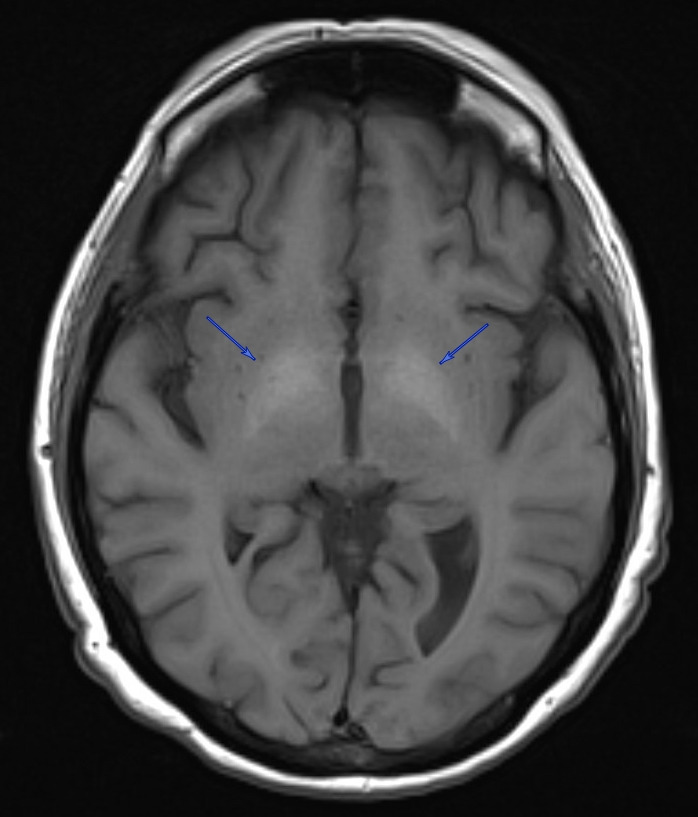
A 50-year-old woman with a history of alcohol use disorder complicated by alcoholic cirrhosis, hepatic encephalopathy, mild Korsakoff syndrome, variceal bleeds, opioid use disorder with chronic pain, attention deficit hyperactivity disorder, major depressive disorder, and hallucinations presented from an alcohol detoxification program with worsening confusion and hallucinations, as well as suffering an unwitnessed fall. Vitals on admission were all stable and within normal limits. The physical exam was largely unremarkable, with the patient oriented only to self and location, and an otherwise intact neurological exam. Notably, gait was not evaluated at the time of admission due to the patient’s high risk of falls. History was limited mainly due to confusion and visual hallucinations. Her pertinent labs at that time showed hemoglobin 10.0 g/dL (normal 11.2 – 14.9 g/dL), MCV 78.4 fL (normal 85.2 – 100.2 fL), ammonia 52 umol/L (normal 1 – 39 umol/L), iron 16 UG/DL (normal 37 – 170 UG/DL), ferritin 18 NG/ML (normal 5 – 204 NG/ML), transferrin saturation 4% (normal 15 – 50%), and ethanol 258 MG/DL (normal 0 MG/DL). Venous blood gas and thyroid-stimulating hormone levels were within normal limits, and urinalysis was negative for signs of infection. CT angiography of the head showed no intracranial abnormalities. She was empirically treated for alcohol withdrawal, hepatic encephalopathy, and Korsakoff syndrome. She was also evaluated by psychiatry for agitation and was started on olanzapine as needed. After a week of admission and resolution of hallucinations and return of baseline mental status, the patient was discharged to a rehab facility, from which she was readmitted a week after for acute encephalopathy; she had been started on amitriptyline at the rehab facility, which was suspected to have caused anticholinergic delirium. Amitriptyline was discontinued, and she was initiated on aripiprazole leading to improvements in hallucinations and mental status. On chart review, a prior MRI from 2 months earlier revealed T1-hyperintensities in the bilateral globi pallidi, consistent with manganese accumulation in a setting of a portosystemic shunt and chronic hepatic dysfunction due to alcohol abuse (Figure 1). Manganese level was 14.4 ug/L (normal 4.2 – 16.5 ug/L). Copper, lead, ceruloplasmin, and carboxyhemoglobin were all within normal limits. The patient was also evaluated by neurology, but despite the imaging findings, she exhibited no clinical features of Parkinsonism, and neurological evaluations remained unrevealing throughout her multiple admissions.

Manganese is a trace element essential for various biological functions, including metabolism, detoxification, immune system production, bone development, and neurodevelopment. Naturally, there remain optimal concentrations, and the body must maintain homeostasis to avoid sequelae of toxicity.1,2 Excess manganese can accumulate in the basal ganglia, namely the globus pallidus, and damage dopamine transporters and receptors, as well as inducing oxidative stress and impairing glial cell function.3,4 Historically, manganese neurotoxicity was first identified in miners and nearby residents exposed to manganese ore and dust, who developed psychiatric and cognitive disturbances, as well as Parkinsonian features such as bradykinesia, rigidity, and tremor.5–7 In the general population, however, the primary source of manganese is drinking water, where it typically coexists with iron. Because iron and manganese utilize the same transport proteins, iron deficiency can contribute to increased manganese absorption. Manganese is generally excreted in bile, so individuals with liver dysfunction can also develop toxic levels of the element.8
Manganese toxicity is classically associated with a psychiatric and cognitive disturbances and a parkinsonian phenotype such as bradykinesia, cogwheel rigidity, and postural tremors.9 However, it differs from idiopathic Parkinson’s disease in several ways, such as reduced resting tremor, the absence of Lewy bodies, and a limited response to levodopa.10,11 While this disease presentation was first discovered in individuals with high levels of environmental manganese exposure, the literature has demonstrated similar findings in individuals with liver dysfunction. Hepatic and biliary clearance are crucial to manganese homeostasis, and cases of chronic hepatic cirrhosis have been reported to develop elevated blood manganese concentrations, as well as mild cognitive impairments, postural tremors, and MRI findings of hyperintensity in the basal ganglia.12–15 These MRI findings have been uniquely associated with chronic liver failure and manganese: a study of cirrhotic patients comparing manganese levels to other metals, including copper, mercury, cadmium, zinc, arsenic, iron, and lead, confirmed that manganese accumulation within the pallidi was the cause of the T1 hyperintensities in the globi pallidi.16 Patients who developed cognitive and extrapyramidal changes were those with imaging findings, and post-mortem analysis showed pallidal manganese concentrations 4.7 times higher than liver failure patients without imaging findings.
It is of significance that the patient discussed in this report exhibited a range of emotional disturbance, compulsive behavior, hallucinations, and memory loss, but did not show any signs of motor deficits typically expected with Parkinsonism. Given our patient’s manganese level of 14.4 ug/L, her symptoms coincide with studies that have reported an association with cognitive decline in patients within the upper quartile of blood manganese levels (>11.18 ug/L).17 A manganese of 14.4 ug/L is within the upper limits of normal as per the Rhode Island Hospital laboratory criteria (normal 4.2 – 16.5 ug/L), thus the case is unique in that the patient exhibited T1 hyperintensities in globi pallidi despite such manganese levels, as other cases with similar imaging findings have reported levels ranging from 20 ug/L to 195 ug/L.18–20 This suggests the possible conclusion that manganese neurotoxicity may be achieved with concentrations lower than what would typically be defined as abnormal, and clinicians must remain vigilant with similar patient populations for potential sequelae despite “normal” metal levels. Additionally, studies of patients with liver failure have reported that while all those who demonstrated Parkinsonism had the characteristic T1 signal abnormalities, not all those with the key imaging findings necessarily demonstrated features of Parkinsonism. However, the medical literature has documented cases of patients with manganese toxicity developing Parkinsonism after a variable latency period during which they exhibit psychiatric disturbances, thus our patient could potentially demonstrate a similar biphasic clinical course, further indicating a need for close follow-up of chronic liver disease patients from a neurologic perspective.21
Currently, there is limited literature on the prevention and treatment of manganese toxicity; however, concurrent management of iron deficiency may be beneficial. Low iron stores have been shown to increase blood concentrations of divalent metals, such as manganese, with increased manganese accumulation in the brain as a response to iron deficiency. Consequently, restoration of iron stores may, in turn, limit manganese toxicity.22–24 Future management of similar cases will likely benefit from consideration of this potential biphasic presentation of symptoms as well as association with iron deficiency, a very common finding in patients with cirrhosis. This case highlights manganese neurotoxicity as a possible but underrecognized contributor to neuropsychiatric symptoms in patients with hepatic dysfunction. Despite normal manganese levels, MRI findings can show basal ganglia accumulation even in the absence of Parkinsonian motor features, and clinicians must remain vigilant in early identification, which could help anticipate future motor decline and inform management strategies.
Disclosures/Conflicts of Interest
The authors have no conflicts of interest to disclose.
Corresponding author
Kang Woo Kim, MD
Department of Internal Medicine, Brown University Health
Mailing Address: 593 Eddy Street, Providence, RI 02903
Phone: 401-457-3336
Fax: 401-525-2549
Email: kang_woo_kim@brown.edu