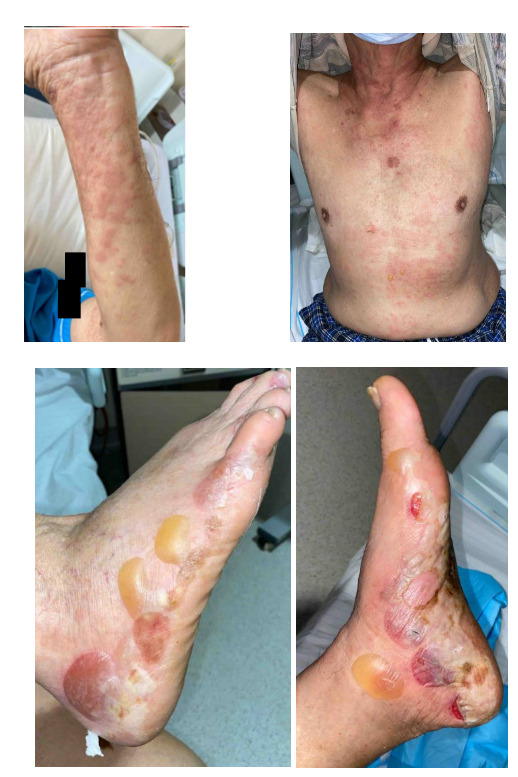
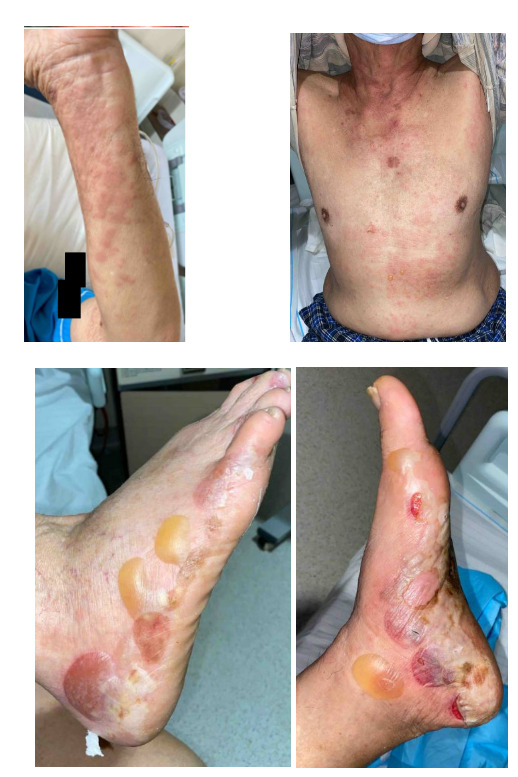
A 79-year-old gentleman, with no significant medical history or family history of skin or autoimmune conditions, presented to the hospital with a 3-week history of persistent, itchy “hives” over his trunk and limbs, on a background of generalized pruritus for 4 months. He also described intermittent episodes of angioedema of his eyes that self-resolve. His general practitioner treated him with low-moderate potency topical corticosteroids, antihistamines, and short courses of low-dose prednisolone 10 mg/day with minimal improvement. Over the week before admission, he noticed several blisters and bullae forming on his feet. On examination, there were generalised urticated plaques observed over the trunk, abdomen, and upper limbs, with two tiny blisters noted over the anterior abdominal wall. However, several large blisters and tense bullae were evident over the bilateral feet, some of which had spontaneously burst (Figure 1). There was no oral or genital mucosal involvement. Nikolsky’s sign was negative.
Initial laboratory tests revealed peripheral eosinophilia (AEC 2.33 x 109/L; reference range 0.05-0.5 x 109/L) with normal renal and liver function. Serum bullous pemphigoid (BP) 180/230 IgG antibodies were negative (reference range < 20 RU/ml) on ELISA testing, but serum indirect immunofluorescence (IIF) anti-basement membrane zone (BMZ) IgG antibodies were detected on both primate oesophagus and salt-split skin substrate, demonstrating an epidermal pattern on salt-split skin. As the patient was not keen to proceed with a skin biopsy, he was treated for BP, based on the clinical findings and serum IIF results, with oral prednisolone 40 mg OM (between 0.5-0.75 mg/kg range), high-dose antihistamines (telfast 360 mg BD), and high-potency topical corticosteroids (clobetasol dipropionate 0.05% cream BD). Subsequently, doxycycline 100 mg BD and eventually azathioprine 50 mg OM were added due to persistent blisters. Thereafter, the patient had a good clinical response, with complete resolution of blisters and skin erosions.
Urticaria refers to the development of classical wheals and/or angioedema, with or without identifiable triggers.1 It is further split into acute (< 6 weeks) and chronic (> 6 weeks) urticaria, based on symptom duration.1 For persistent or recurrent urticated lesions, it is always important to distinguish between common urticaria and atypical lesions that may suggest dermatological mimics or systemic urticarial syndromes.2 In general, typical urticaria is characterized by elevated, erythematous, edematous, well-circumscribed plaques with central pallor, each lesion resolving spontaneously within 24-36 hours, with no residual scarring or hyperpigmentation.2 On the contrary, atypical “wheals” may have infiltrated plaques, persistent lesions > 24-36h, post-inflammatory hyperpigmentation or residual scarring, as well as secondary skin lesions such as papules, vesicles, blisters/bullae, scales, and crusts.2 In such cases, diagnostic considerations could include urticarial vasculitis, urticarial dermatitis, autoimmune blistering diseases, cutaneous mastocytosis, or systemic urticarial syndromes associated with connective tissue or autoinflammatory diseases. In our patient, although he had persistent urticated lesions, the eventual development of secondary skin lesions, including blisters and bullae, suggested an autoimmune blistering disease such as bullous pemphigoid or paraneoplastic pemphigoid. Dermatologic emergencies such as Stevens-Johnson syndrome/toxic epidermal necrolysis were also considered but deemed unlikely given the clinical presentation, lack of mucosal involvement, negative Nikolsky’s sign, and absence of identifiable inciting drug/infective triggers.
Bullous pemphigoid is a subepidermal, autoimmune blistering disease, commonly seen in older adults > 65 years of age, characterised by autoantibodies targeting the basement membrane zone, with major target antigens BP 230 and BP 180.3 While the majority of patients develop tense blisters and bullae that are suggestive of immunobullous disease, there is an often misdiagnosed pre-bullous phase of BP found in up to 20% of patients that can last for several weeks to months.4 Clinical manifestations in this stage can be non-specific, ranging from intense pruritus with no obvious skin lesions to urticarial, eczematous, or papular skin rashes lasting for several weeks to months.3–6 Eventually, these patients will develop characteristic tense bullae and vesicles on urticated or erythematous bases, especially over flexural areas.6,7 It is therefore suggested that older adult patients with urticaria or pruritic skin lesions that appear refractory to standard symptomatic treatment should prompt consideration of BP as a differential diagnosis.4 The exact pathophysiology of urticated lesions in BP is not entirely known, but is postulated to be related to IgE-mediated mechanisms.7 In particular, the presence of IgE autoantibodies against COL17 (a basement membrane antigen) has been reported in many BP patients, and seems to portend greater disease severity and treatment refractoriness.8 Other studies have even suggested that it may be useful to detect pre-bullous/early BP.9 However, ELISA assays for anti-BP180 IgE are not widely available, and further studies have shown that they provide only marginal benefit as compared to standard assays.10
Traditionally, skin biopsy is the gold standard for diagnosing BP by demonstrating linear IgG and/or C3 deposits along the dermal-epidermal junction of perilesional skin under direct immunofluorescence (DIF).6 The combination of a characteristic bullous eruption in an older adult with suggestive DIF findings confers high diagnostic sensitivity (90%) and specificity (83%).6 Nowadays, two other non-invasive laboratory techniques are increasingly employed for the diagnosis of BP, which include serum cutaneous indirect immunofluorescence (IIF) for BMZ IgG antibodies, epidermal or epidermal-dermal antibodies with salt-split skin, or BMZ IgG antibodies with monkey oesophagus substrate, and serum ELISA testing for BP 180/230 IgG autoantibodies.11 However, these tests generally have lower sensitivity than DIF from the skin biopsy sample,6 where, for instance, the monkey oesophagus substrate expresses low levels of BP180 protein, thereby limiting the detection of BMZ IgG antibodies.12 In our patient, his clinical findings and serum IIF supported the diagnosis of BP, and hence a trial of immunomodulatory therapy with careful monitoring of treatment response was reasonable given that he was not keen on a skin biopsy. However, the diagnostic limitation of such an approach had to be conveyed to the patient, where false positive serum IIF could rarely be caused by technical factors including substrate cross-reactivity, and anti-BP180/230 antibodies could be found in other subepithelial immunobullous diseases such as linear IgA disease, and mucous membrane pemphigoid.12 Our patient had discrepant findings with positive IIF BMZ IgG antibodies but negative ELISA testing for anti-BP180/230 antibodies. This could be attributed to the lack of sensitivity in commercially available ELISA assays that is not superior to IIF studies with salt-split skin, which may be attributed to antigen epitope variation6 or lack of circulating antibodies in certain atypical BP presentations.13 This may be improved with expansion of ELISA antibody detection with incorporation of NC16A domain and other extracellular areas of BP180/230 antigens, or usage of traditional immunoblotting/immunoprecipitation techniques, although these are either not widely available or technically demanding and time-consuming.6 On the other hand, for patients who have negative serum IIF and/or ELISA testing, skin biopsy should be performed to confirm the clinical diagnosis of BP.
Depending on the extent/severity of disease, initial treatment generally involves a combination of high-potency topical corticosteroids, oral corticosteroids (low-moderate dose prednisolone) or immunomodulatory drugs such as dapsone to induce remission.5 Other immunosuppressive agents, including azathioprine, mycophenolate mofetil, and biologics, can be considered as steroid-sparers subsequently.5 In summary, our patient presented with a 3-week history of refractory urticaria on a background of chronic generalised pruritus and was eventually diagnosed and treated for BP. Failure to consider this entity in older adult patients with persistent urticaria/itch refractory to standard antihistamine treatment may lead to delayed diagnosis, functional impairment, and poor quality of life.
Disclosures/Conflicts of Interest
The authors have no conflicts of interest to disclose.
Corresponding author
Dr. Isaac KS Ng, MBBS, MRCP (UK)
Senior Resident, Division of Rheumatology and Allergy,
Department of Medicine,
National University Hospital, Singapore
Singapore 119074
Email: isaac.ngks@mohh.com.sg