A 76-year-old man with a pertinent history of frontotemporal dementia presented to the hospital from his memory care unit with an episode of unresponsiveness in the setting of subacute behavioral dysregulation in the form of worsening agitation. There was no history of fevers, antecedent head trauma, or recent medication adjustments. On admission, his labs, including basic metabolic panel, complete blood count, and urine analysis, were unremarkable. TSH was low at 0.108 (ref: 0.330-4.120 uIU/ml), but Total T3 and Free T4 were normal. Treponemal IgM and IgG were unreactive.
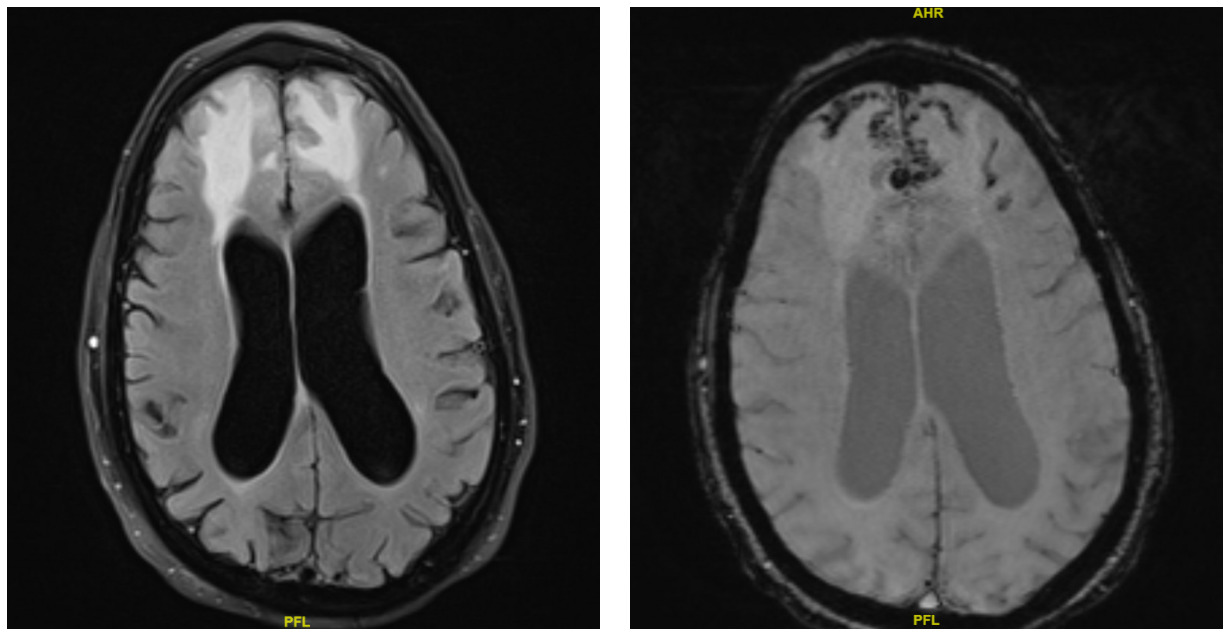
A CT brain was performed in the emergency department, which showed vasogenic edema in the bilateral frontal lobes with trace subarachnoid hemorrhage and hemorrhagic contusion in the right frontal lobe. A repeat CT performed six hours later showed no interval change. The patient was subsequently admitted, and both neurosurgery and neurology were consulted. They recommended initiating IV dexamethasone for vasogenic edema and IV levetiracetam for seizure prophylaxis. A routine EEG showed diffuse theta background slowing, suggesting an underlying mild to moderate diffuse encephalopathy without focal epileptiform discharges. An MRI of the brain was performed, which showed bifrontal vasogenic edema with both superficial siderosis and microhemorrhages, consistent with cerebral amyloid angiopathy-related inflammation (CAA-RI) (Figure 1).
_and_swi_imaging_showi.png)
The patient was initiated on treatment with high-dose IV methylprednisolone 1g/day for 5 days. His hospital course was complicated by agitation, which was treated with reorientation and as-needed anti-psychotics. The agitation was felt to be multifactorial secondary to his underlying acute process (CAA-RI), his underlying frontotemporal dementia, medication side effect due to high-dose steroids, and hospital-acquired delirium. His mental status improved throughout his stay, and on the day of discharge, the patient had returned to his baseline mental status. He was discharged with a prolonged steroid taper and follow-up with his primary neurologist.
Patients with preexisting cognitive impairment can experience a decline in their usual mental status for multiple reasons. Common causes include systemic or central nervous system infections, inflammatory conditions that trigger cytokine release (e.g., interleukins), cerebrovascular events (both ischemic and hemorrhagic), new-onset seizures, CNS medication side effects, metabolic or electrolyte disturbances, and progression of the underlying cognitive disorder. Evaluation should include basic laboratory studies, brain imaging (preferably MRI), EEG, and cerebrospinal fluid analysis when appropriate. It is essential to rule out reversible causes before attributing cognitive changes to disease progression. When reversible conditions are identified, they must be promptly recognized and treated to prevent further deterioration in mental status.
Cerebral amyloid angiopathy–related inflammation (CAA-RI) is an uncommon yet aggressive variant of CAA, characterized by diverse clinical and imaging features. First reported by Reid and Maloney in 1974, it typically presents with an acute or subacute decline in cognition or behavioral changes. Additional manifestations may include seizures, headaches, aphasia, encephalopathy, and generalized weakness. Pathologically, CAA-RI is divided into two forms: a non-destructive perivascular inflammation known as inflammatory CAA, and a transmural or intramural inflammatory process termed Aβ-related angiitis (ABRA). Although brain biopsy or autopsy remains the diagnostic gold standard, these procedures are invasive, prompting the development of clinical and radiologic diagnostic criteria.1 In 2011, Chung et al. introduced the Boston criteria based on combined clinical and imaging findings.2 Auriel et al. later revised these criteria in 2016, highlighting specific white-matter hyperintensity patterns to differentiate probable from possible CAA-RI and incorporating cortical superficial siderosis as an indicator of hemorrhage.3
In patients who meet the criteria for probable CAA-RI, empiric immunosuppressive therapy may be initiated to avoid the need for brain biopsy. If there is no clinical or radiographic improvement after approximately three weeks of treatment, a biopsy should then be considered. The presence of patchy or confluent T2 hyperintensities on MRI, along with strictly lobar microbleeds or cortical superficial siderosis on susceptibility-weighted imaging, supports the diagnosis of CAA-RI. MRI findings of T2 white-matter hyperintensities, combined with pathological evidence of vascular Aβ-associated inflammation, further reinforce the diagnosis.4
Most patients with CAA-RI demonstrate elevated CSF protein and pleocytosis, typically without oligoclonal bands. In some cases, anti-Aβ antibodies have been detected in the CSF, and their levels tend to decline with immunosuppressive treatment. The differential diagnosis includes several vascular and inflammatory disorders, such as posterior reversible encephalopathy syndrome (PRES), primary angiitis of the central nervous system (PACNS), primary CNS lymphoma, and autoimmune encephalitis. As illustrated in this patient, it is crucial to consider CAA-RI early, establish the diagnosis promptly, and initiate appropriate therapy. Unlike classic CAA, which currently lacks effective treatment, CAA-RI often responds to systemic corticosteroids, with or without additional immunosuppressive agents, leading to improvement in both clinical symptoms and imaging abnormalities. Importantly, brain MRA and cerebral angiography have limited sensitivity for detecting CAA-RI due to the involvement of small-caliber vessels. Therefore, clinicians must be well-informed about this condition and obtain appropriate MRI studies when clinically warranted.5,6
Disclosures/Conflicts of Interest
None
Corresponding author
Edward C Daingerfield, MD
The Miriam Hospital
Warren Alpert Medical School of Brown University
Providence, RI, 02906
Email: edaingerfield@brownhealth.org