Abbreviations
MCD: minimal change disease
FSGS: focal segmental glomerulosclerosis
PLEX: plasmapheresis
AKI: acute kidney injury
Immunoglobulin G: IgG
Background
Nephrotic syndrome, which is characterized by significant proteinuria secondary to defects in the glomerular filtration barrier, commonly manifests in patients as hypoalbuminemia and periorbital and/or peripheral edema.1 Minimal change disease (MCD) and focal segmental glomerulosclerosis (FSGS) are two common causes of nephrotic syndrome that are caused by injury to the renal podocytes. While historically described as distinct disease processes, an expanding body of clinical and translational evidence over the last decade supports a continuum of pathology between MCD and immune-mediated FSGS, with MCD representing an early stage of this continuum.2 The recent identification of anti-nephrin antibodies, which are associated with both MCD and immune-mediated FSGS, further supports this continuum and may link the two conditions pathophysiologically.2,3
Case Presentation
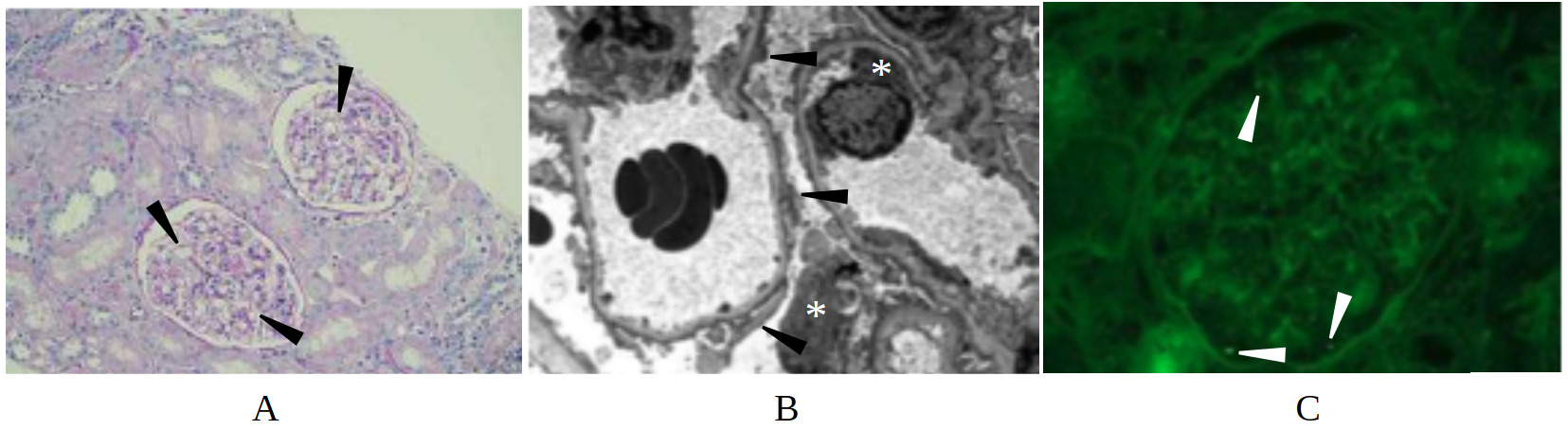
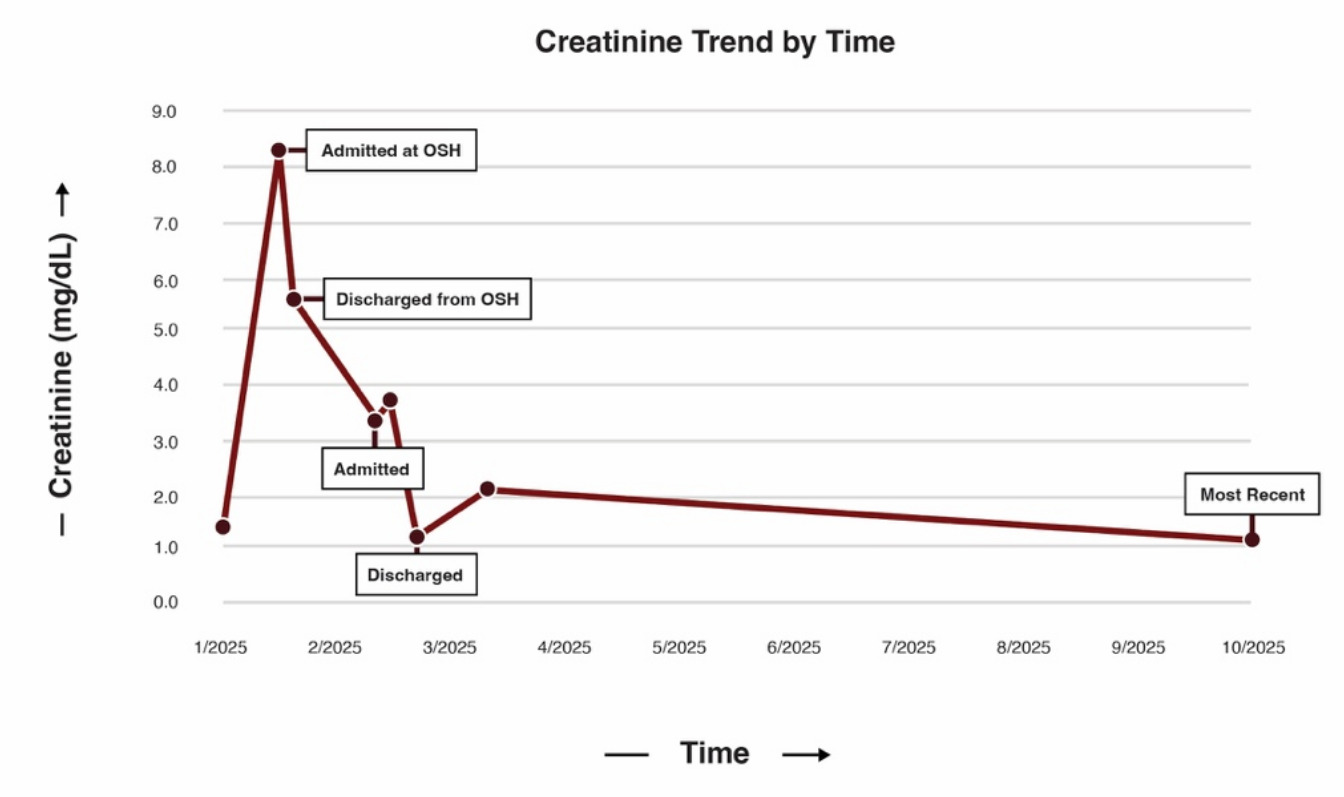
A 32-year-old female with a known history of MCD presented to the emergency department at the recommendation of her outpatient nephrologist with symptoms of volume overload, worsening renal function, and emesis. She was diagnosed with MCD via renal biopsy seven months prior (Figure 1) and was initially maintained on furosemide and dual immunosuppressant therapy of cyclosporine and prednisone (down-titrated to 20 mg daily due to prior complications of steroid-induced psychosis and hallucinations). A month prior to admission, she received care at an outside hospital for acute kidney injury (AKI) in the setting of volume overload with a serum creatinine of 8.0 mg/dL on admission (baseline 0.80 mg/dL, reference 0.5-1.2 mg/dL). The AKI was thought to be multifactorial secondary to recent trimethoprim/sulfamethoxazole use for a finger infection and pre-renal injury in the setting of persistent emesis and furosemide use. Both cyclosporine and furosemide were discontinued during that hospitalization, and she was discharged on prednisone monotherapy at 40 mg daily. Although her renal function initially improved and creatinine levels stabilized at 3.30 mg/dL, the patient gradually developed recurrent anasarca. This edema was refractory to both escalation of prednisone to 60 mg daily and reinitiation of furosemide at 10 mg daily per outpatient nephrology, prompting the current hospital admission.

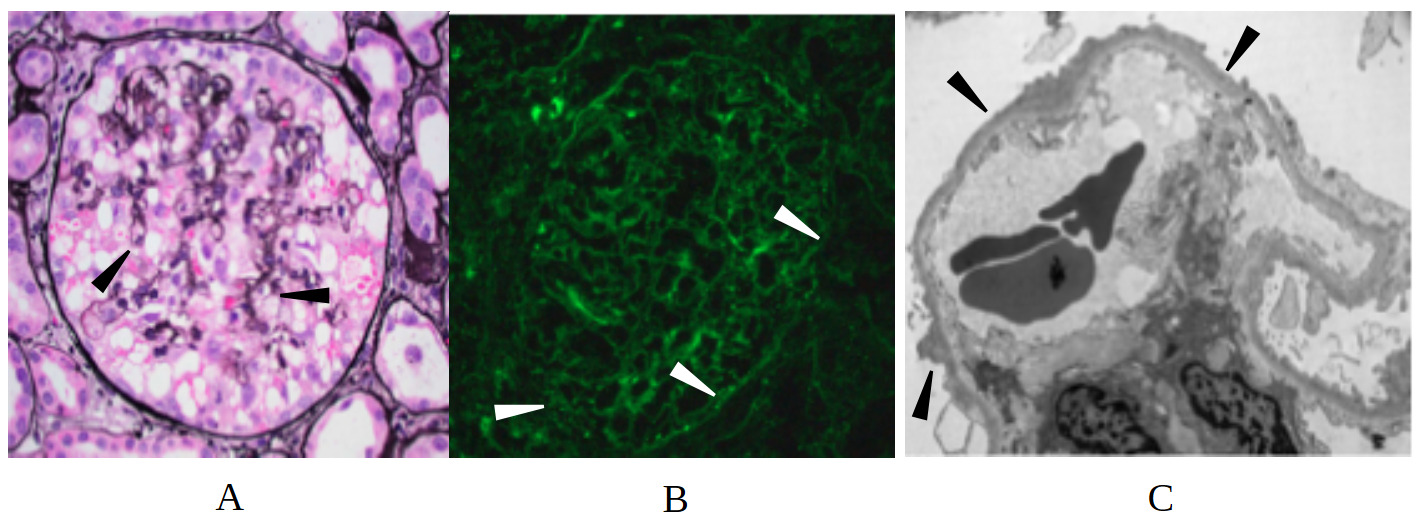
On arrival, the patient’s vital signs were remarkable for hypertension (169/114 mmHg) and physical exam was notable for bibasilar crackles on pulmonary auscultation, periorbital edema, 2+ pitting edema of the upper extremities, and 4+ pitting edema in the bilateral lower extremities. Laboratory evaluation revealed elevated serum creatinine (3.37 mg/dL), elevated blood urea nitrogen level (92 mg/dL, reference 7-20 mg/dL), hypoalbuminemia (1.5 g/dL, reference 3.4-5.4 g/dL), and hyperphosphatemia (6.0 mg/dL, reference 2.5-4.5 mg/dL). Urinalysis demonstrated 4+ proteinuria, 4+ glucosuria, 12 red blood cells per high-powered field, and 6-10 hyaline casts per low-powered field. There was no evidence of dysmorphic red blood cells, acanthocytes, or red blood cell casts. 24-hour urine collection was notable for over 40 g of protein with adequate urine collection (reference < 150 mg/day), and urine protein/creatinine 32.1 (reference < 0.2). Broad serologic workup including ANCA, ANA, dsDNA, MPO, C3, C4, Hepatitis B/C antibodies, HIV, RPR, APOL1, PLA2-R, parvovirus, EBV, and CMV was unremarkable. Serologic evaluation was notable only for positive anti–Sjögren’s-syndrome-related antigen A autoantibodies, and subsequent rheumatologic evaluation with Schirmer’s test and parotid ultrasound were overall equivocal for Sjogren’s syndrome. The patient was started on IV albumin 25 g every 8 hours, which was continued for 48 hours with no response in serum creatinine (3.37 to 3.53 mg/dL). On day 2 of the hospitalization, a repeat renal biopsy revealed extensive podocyte foot process effacement, focal glomerulosclerosis, patchy tubulointerstitial inflammation, and punctate IgG podocyte cytoplasmic staining without evidence for an immune mediated process (Figure 2). This constellation of findings was consistent with the collapsing variant of FSGS.

The patient was subsequently started on prednisone 60 mg daily and plasmapheresis (PLEX) for seven days. Additionally, her diuretics were increased to bumetanide 2 mg twice daily for better diuresis. The patient showed a significant response to the modified treatment regimen, both in clinical and laboratory parameters. Following seven days of PLEX and diuresis, her serum creatinine improved significantly to 1.2 mg/dL with a net fluid loss of 10 liters. She was discharged on prednisone 40 mg daily and atovaquone for Pneumocystis jirovecii prophylaxis, with a plan to follow up with nephrology outpatient.
One month after initial presentation, the patient was readmitted with progressive anasarca despite escalating diuretic dose outpatient. At the time of admission, her creatinine was 1.55 mg/dL, approximately at her new baseline. While admitted and at the advice of her outpatient nephrologist, she was started on rituximab and successfully diuresed with removal of 14 liters and 30 pounds total. She has since completed her second rituximab infusion and continues on prednisone 40 mg with a plan for gradual taper. The patient responded well to therapy, as evidenced by improvements in her serum creatinine (most recently 1.11 mg/dL; Figure 3), proteinuria (urine protein/creatinine 4.1), and anasarca.

Discussion
Focal segmental glomerulosclerosis (FSGS) represents a histopathological diagnosis with diverse etiologies rather than a single disease entity. The traditional binary of “primary” versus “secondary” FSGS is being redefined in light of genetic, immunologic, and hemodynamic factors that influence pathogenesis and therapeutic responsiveness. Our case highlights this spectrum of histopathological disease and identifies potential alternative treatment approaches for refractory disease.
Our patient’s initial presentation appeared to be due to worsening of MCD secondary to pre-renal AKI, though repeat renal biopsy demonstrated findings consistent with collapsing FSGS. The patient’s renal injury and nephrosis were presumed to be immune-mediated, and she was successfully treated with steroids and plasmapheresis followed by rituximab. This presentation is the first described case of pathologic progression from MCD to FSGS successfully treated with dual immunosuppression and plasmapheresis.
Although MCD and FSGS may have similar clinical presentations, historical literature differentiates them based on their histopathology and prognosis. On electron microscopy, MCD is identified by the diffuse effacement of the podocyte foot processes, which challenges the integrity of the filtration barrier and promotes increased urinary protein loss.3 In addition to foot process effacement, FSGS is characterized by focal glomerular scarring caused by the deposition of hyaline material around glomerular vessels and subsequent narrowing of the vasculature.3 Unlike FSGS, MCD tends to have a favorable prognosis due to marked therapeutic response to corticosteroid therapy and minimal risk of progression to end-stage renal disease. Despite these differences, recent literature has explored whether MCD and primary, immune-mediated FSGS lie along a continuum of a single disease process.2 As FSGS is defined by sclerosis in addition to podocyte effacement, this model suggests that persistent immunologic insult to the podocytes and resultant sclerosis results in the histologic transition from MCD to FSGS.2 Steroid-responsive MCD patients, therefore, have limited persistent podocyte injury and do not develop the sclerosis that defines the transition to FSGS.3
De Vriese et al. argue for a paradigm shift: FSGS should be viewed not as a disease but as a histologic endpoint.4 They propose a six-subtype classification that distinguishes primary, adaptive, genetic, virus-associated, drug-induced, and APOL1-associated forms. This classification would not only further support the spectrum of podocyte-related disease but would also help create an algorithmic approach for both diagnosis and treatment, lending Clinicians with more tailored options and the avoidance of unnecessary and ultimately futile treatment regimens. Towards more effective treatment, Bierzynska et al. compiled over 40 gene mutations linked to familial or early-onset steroid-resistant nephrotic syndrome.5 Most monogenic forms are unresponsive to immunosuppressive therapy, highlighting the need for early genetic testing, particularly in pediatric cases and adults with familial or syndromic features.
While our case is most consistent with histopathologic progression from MCD to FSGS along this continuum, an alternate consideration is the initial misdiagnosis of FSGS due to the biopsy limitations. As FSGS lesions cause selective injury to only a subset of glomeruli, inadvertent sampling of an area with unaffected glomeruli on biopsy can lead to misdiagnosis.3 Additionally, early-stage FSGS may lack its characteristic hyaline deposition and segmental sclerosis and only shows diffuse podocyte effacement on biopsy, making it indistinguishable from MCD. This case reinforces the role of repeat renal biopsy in patients with steroid-refractory MCD, both to identify patients with initially misdiagnosed FSGS and to identify patients who have progressed from MCD to FSGS.
The patient’s initial renal biopsy demonstrated extensive podocyte foot process effacement consistent with MCD, along with immunoglobulin G (IgG) dusting on immunofluorescence, which refers to the deposition of podocyte-associated IgG autoantibodies in a punctate fashion.6 Nephrin is an essential protein that contributes to the structural integrity of the slit diaphragms that bridge adjacent podocytes together; serum IgG autoantibodies that target nephrin thereby allows intravascular proteins within the glomerulus to pathologically leak into the urine. An expanding body of evidence suggests a close association with anti-nephrin autoantibodies and the pathogenesis of MCD and immune-mediated or idiopathic FSGS.2,6–8 In one study by Hengel et al., anti-nephrin autoantibodies were identified in 44% of adults with MCD and 9% of adults with primary FSGS.7 Furthermore, there was a positive association between anti-nephrin levels and disease activity. While IgG anti-nephrin autoantibodies represent one possible pathophysiologic link, non-IgG autoantibodies or alternate biomarkers have also been proposed as drivers of podocyte injury in these conditions.3,7
This case additionally highlights the successful use of PLEX in the treatment of a steroid-resistant primary podocytopathy. Despite treatment with high-dose steroids and adjunctive cyclosporine, significant clinical improvement was observed in our patient only after PLEX was initiated and continued for a 7-day treatment course. In patients with dusting, PLEX facilitates the direct removal of circulating autoantibodies, thereby eliminating the source of podocyte damage. While PLEX to clear anti-nephrin antibodies is well reported in the renal transplant population, this report is among the few that describe successful plasmapheresis in a non-renal transplant patient. In addition to PLEX, emerging research suggests that steroid-resistant nephrotic syndrome may be effectively treated with B-cell-directed therapies, such as rituximab or mycophenolate mofetil, that directly limit the production of IgG anti-nephrin antibodies, thereby preventing recurrence of symptoms.9
To our knowledge, this report is the first published case of MCD progression to the collapsing-variant of FSGS successfully treated with plasmapheresis and dual immunosuppression. This case supports the emerging evidence base for a disease continuum between MCD and immune-mediated FSGS, with particular attention to anti-nephrin antibodies as a driving factor in these podocytopathies. In similar cases of steroid-refractory MCD, early repeat renal biopsy may clarify the true diagnosis, while plasmapheresis can be effectively employed to clear circulating antibodies.
Disclosures/Conflicts of Interest
The authors have no disclosures to report
Corresponding author
Ritika Muthyala,
BS 3500 Camp Bowie Blvd
Fort Worth, TX, 76107
Email: ritikamuthyala@my.unthsc.edu