Background
Peutz-Jeghers syndrome (PJS) is a rare autosomal dominant disorder that presents with multiple gastrointestinal hamartomatous polyps and mucocutaneous hyperpigmentation. These hamartomatous polyps occur most commonly in the small intestine but may affect any part of the gastrointestinal tract.1 It may cause complications like intussusception, malignant transformation of the hamartomas into colorectal cancer, and may also present with occult or overt gastrointestinal bleeding.2 Jehovah’s Witnesses are members of a Christian religious group known for their staunch belief in refusing blood product transfusions even in the face of severe life-threatening anemia. This necessitates that clinicians familiarize themselves with evidence-based transfusion-free anemia management while upholding patients’ religious beliefs and autonomy. Here we report a unique presentation of an adult female who is a Jehovah’s Witness with severe iron deficiency anemia and chronic occult gastrointestinal bleeding who was diagnosed with PJS without any prior family history.
Case presentation
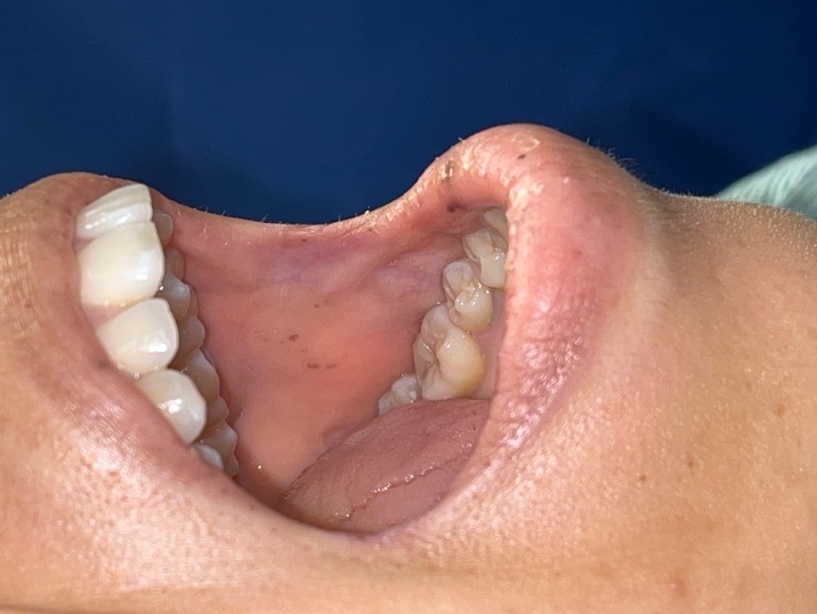
A 29-year-old woman with a past medical history of severe anemia and lactose intolerance presented to the emergency department with complaints of nausea, vomiting, non-bloody diarrhea, and intermittent diffuse abdominal pain for 5 days following the intake of food with cheese. She denied fever, chills, hematuria, hematochezia, melena, or a history of heavy menstrual bleeding. She took ibuprofen 2-3 times every month for chronic headaches and iron supplements one year before presentation for severe anemia. However, she did not have any previous records or workup available for her anemia, except for a complete blood count from a year back showing a hemoglobin level of 6.0 g/dL and low MCV. She was a Jehovah’s Witness and, as per her religious beliefs, she declined blood transfusions. At presentation, she was afebrile, with a blood pressure of 134/78 mmHg, pulse rate of 89/min, and was maintaining normal saturation on ambient air. Physical examination showed severely pale mucosa and hyperpigmented lesions on the buccal mucosa (Figure 1). Laboratory evaluation showed severe iron deficiency anemia with a hemoglobin of 4.8 g/dL, MCV 64fL, serum iron of 12 mcg/dL, TIBC 423 mcg/dL, transferrin saturation of 2.8%, LDH of 238 IU/L, and ferritin of 2.27 ng/mL.

Due to her being a Jehovah’s Witness and the severe anemia that she presented with, the medical team had an extensive discussion with the patient and her family regarding the risk of severe anemia, life-threatening consequences that can occur in case of any acute bleeding, and the importance of transfusion of packed red blood cells in such situations. The team also discussed the alternative options to transfusion that could be offered. After a detailed discussion, she refused to transfusion of blood products, even in the case of any acute life-threatening hemorrhage. An interdisciplinary consultation was held with the transfusion free medicine service, and the decision was made to manage her with intravenous iron dextran and erythropoietin, in addition to supplementation with folic acid, vitamin B12, and ascorbic acid. She was maintained on strict bed rest until her hemoglobin levels improved and received intravenous pantoprazole for gastrointestinal stress ulcer prophylaxis. Beta blockers were not initiated to enhance oxygen utilization, as she was euvolemic and her heart rate remained within the normal range of 70 to 90 beats per minute.
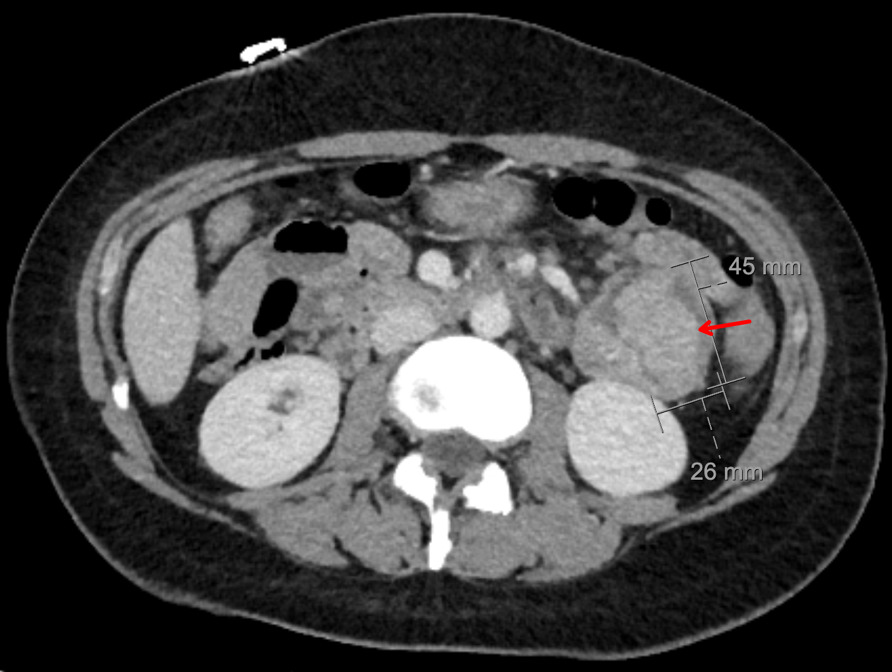
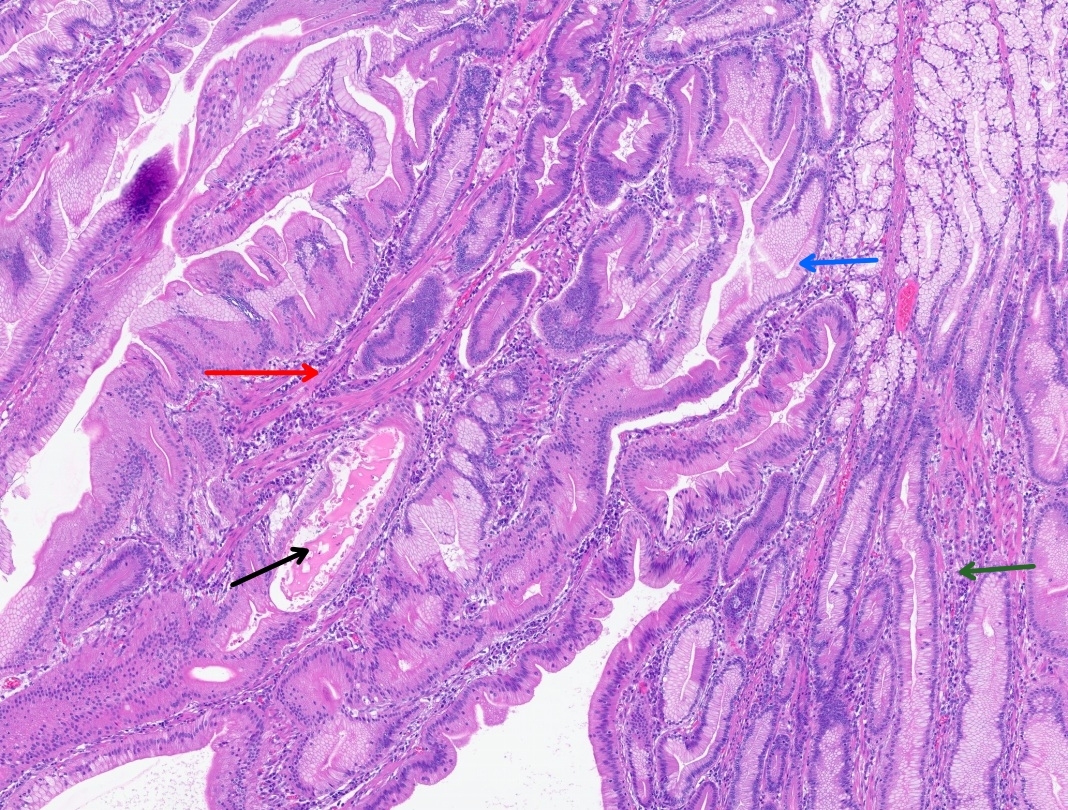
CT abdomen and pelvis with enterography showed numerous intraluminal small polyps in the jejunum largest measuring up to 4.5 x 2.6 cm, 3.0 cm polyp in the left lower abdomen, short segment small bowel intussusception with a polyp as lead point in the right abdomen without associated bowel obstruction or inflammatory bowel thickening, reactive mesenteric lymph nodes and splenomegaly (14.8 cm) (Figure 2). Esophagogastroduodenoscopy and colonoscopy were done, which showed few sessile and semi-pedunculated polyps measuring 4 - 10 mm in the ascending, transverse, descending, and sigmoid colon, multiple large polyps (sessile, semi-pedunculated, and pedunculated) measuring 6 - 20 mm in the rectum, multiple 3 to 8 mm sessile polyps in the gastric fundus and gastric body and a single 15 mm pedunculated polyp at the pylorus. There was no bleeding, and no stigmata of recent bleeding. Polypectomy of a pyloric polyp was done, and histopathology showed features of a hamartomatous polyp with arborizing compact bundles of smooth muscle (Figure 3). Genetic testing was positive for a heterozygous pathogenic variant [c.256C>T (p.R86*)] in the Serine/threonine kinase 11 (STK11/LKB1) gene, establishing the diagnosis of Peutz-Jeghers syndrome (PJS). She mentioned that her parents had screening colonoscopies, which were normal, and denied any family history of PJS. After 5 days of hospitalization, her hemoglobin level remained stable, and there were no overt signs of bleeding. Hence, she was discharged on ferrous fumarate 325mg every other day, ascorbic acid 500 mg twice daily, and folic acid 1 mg daily. Her hemoglobin at discharge was 5.0 g/dL, improved to 10.9 g/dL at 4 weeks, and to 14.0 g/dL at the 4-month follow-up.

_lined_by_fo.jpg)
Since PJS patients are at increased risk of both gastrointestinal and extragastrointestinal cancers, she underwent comprehensive multimodal imaging for malignancy surveillance on follow-up, including bilateral breast magnetic resonance imaging (MRI), MRI of the chest, abdomen, and pelvis. These studies were negative for any malignancy of the breast, bowel, pancreas, gynecologic organs, or other solid organs. After 8 months since diagnosis, the patient underwent robotic-assisted laparoscopic polypectomy of her jejunum. She remained free of symptoms at subsequent follow-up.
Discussion
Peutz-Jeghers syndrome (PJS) is a rare autosomal dominant inherited disorder, but may occur sporadically in up to 30% cases, as in our patient, with no prior family history.3 It has an estimated incidence ranging between 1 in 8,300 and 1 in 200,000 individuals.3 Despite autosomal dominance, it may have variable penetrance, leading to clinical heterogeneity and a challenge in diagnosis. A clinical diagnosis of PJS is made when any one of the following is present: (i) two or more histologically confirmed PJS polyps, (ii) any number of PJS polyps with a family history of PJS in a close relative, (iii) characteristic mucocutaneous pigmentation with a family history of PJS, or (iv) any number of PJS polyps with characteristic mucocutaneous pigmentation.4 94% of those with a clinical diagnosis have a culprit mutation in the serine-threonine kinase 11 (STK11/LKB1) tumor suppressor gene located on chromosome 19.4 This mutation leads to dysregulated growth, forming hamartomatous polyps, and increasing the risk of malignancy.1 Our patient had more than two histologically confirmed hamartomatous polyps, characteristic mucocutaneous pigmentation, and a mutation in the STK11 gene, confirming the diagnosis of PJS. The mucocutaneous pigmented lesions seen in 95% of patients with PJS commonly occur in the lips, buccal mucosa, peri-anal region, hands, and feet.4 Polyps of varying sizes and numbers most commonly involve the small intestine (60-90%), but may also be present in the colon (50-64%), stomach (49%), and the rectum (32%).1,4 Polyps have also been reported at extraintestinal sites like the gallbladder, bronchi, bladder, and ureter.4 These polyps lead to various complications, including intussusception and small bowel obstruction, chronic occult gastrointestinal bleeding causing severe iron deficiency anemia, as in our case, or malignant transformation.
Management of chronic occult gastrointestinal bleeding in our patient was complicated due to her religious beliefs as a Jehovah’s Witness. Jehovah’s Witnesses is a Christian denomination whose religious beliefs preclude them from receiving any blood product transfusion. Managing such patients with severe anemia presents a medical and ethical challenge where a physician needs to provide evidence-based care with bloodless medicine while simultaneously upholding patients’ religious beliefs and autonomy. We treated her with intravenous iron, an erythropoiesis-stimulating agent (ESA), and vitamin supplements following the “Transfusion-free anemia management” protocol in our hospital for patients who refuse allogenic blood transfusions (Table 1). Studies have shown that in severe anemia, a combination of erythropoietin (short half-life) and darbepoetin (longer half-life) produces a rapid and sustained erythropoietic response compared with either agent alone.5 Iron therapy is also recommended along with ESA to improve erythropoiesis. In our case, the patient received 1 gram of IV iron dextran during hospitalization and was discharged on a daily oral iron regimen. However, some studies have shown that alternate-day oral iron therapy is more beneficial as it lowers hepcidin levels, thus improving iron absorption, while also reducing gastrointestinal side effects.5 Ascorbic acid acts as an effective adjuvant in treating severe anemia by enhancing iron absorption and mobilizing stored iron (from ferritin). It reduces functional iron deficiency by increasing iron availability for erythropoiesis, which helps improve responsiveness to ESA, particularly in inflammatory states such as chronic kidney disease or malignancy.6 Folate and vitamin B12 are supplemented since purine synthesis depends on folate and vitamin B12 availability, and deficiency may limit erythropoiesis. Other supportive measures include minimizing iatrogenic blood loss by limiting phlebotomy, using pediatric tubes, and improving oxygen delivery to the tissues. To optimize oxygenation, low-dose non-selective beta blockers (e.g., propranolol) may be used to reduce the heart rate and myocardial oxygen demand, and block the peripheral conversion of T4 to T3, reducing metabolic demands. However, inadvertent use of higher doses may reduce cardiac output and lead to hypoxia of end-organ tissues.7 In case of life-threatening bleeding, four-factor prothrombin complex concentrate (PCC), such as KCentra, containing clotting factors II, VII, IX, and X, may be used.8
PJS patients are at increased risk of both gastrointestinal and extragastrointestinal cancers, most commonly colorectal, breast, small bowel, gastric, and pancreatic cancer, with a 93% lifetime risk of any cancer.1,9 The index cancer usually presents at a mean age of 41 years.3 Hence, for these patients, periodic screening for cancer is of utmost importance. The screening recommendations from various studies, including those outlined in the National Comprehensive Cancer Network guidelines, are presented in Table 2.3,9
Small bowel polypectomy is recommended for all symptomatic polyps and polyps larger than 1 cm in size, to prevent polyp-related mechanical or malignant complications.10 Device-assisted enteroscopy and, if required, surgery-assisted enteroscopy are the standard of care, as was done in our patient with large jejunal polyps, signs of intussusception, and occult bleeding. Surgical resection of the involved bowel segment is reserved for challenging cases.10 All female patients with PJS planning pregnancy should undergo prenatal counseling and genetic testing. Psychological and social support is also very important for PJS patients as they are often diagnosed early in life, face significant emotional burden, and have heightened risk of cancer.
Conclusion
Our case highlights the importance of investigating for PJS in a young adult patient presenting with chronic iron deficiency anemia and classic mucocutaneous hyperpigmented lesions without any pertinent family history and de novo STK 11 mutation. These patients often require a thorough gastrointestinal workup with endoscopy, colonoscopy, and gastrointestinal imaging. Preventing and treating complications like anemia, intussusception, comprehensive cancer surveillance, and polypectomy form the cornerstone of management for such patients. This often requires a multidisciplinary approach and teamwork with diligent counseling and follow-up of PJS patients. We also highlight the challenges of managing severe iron deficiency anemia in patients who are Jehovah’s Witnesses.
Disclosures/Conflicts of Interest
The authors have no conflicts of interest to disclose.
Corresponding author
Virali Gulla, MD
Department of Internal Medicine
Texas Tech University Health Sciences Center, El Paso, TX, USA
Email: vigulla@ttuhsc.edu