Background
Thrombotic microangiopathies (TMA) comprise syndromes characterized by microangiopathic hemolytic anemia, consumptive thrombocytopenia, and organ injury due to microvascular thrombosis.1 TMA may be primary (e.g., severe ADAMTS13 deficiency) or secondary to conditions such as infection, autoimmune disease, medications/toxins, pregnancy, and malignant hypertension. In hypertensive emergencies, acute end-organ injury distinguishes emergencies from urgencies.2
Cocaine can precipitate hypertensive emergency through sympathetic stimulation and vasoconstriction and has been associated with endothelial injury and prothrombotic effects (e.g., platelet activation, increased von Willebrand factor, increased fibrinogen, and impaired endothelial nitric oxide signaling).3–5 Cocaine exposure, malignant hypertension, and complement activation can coexist; complement activation may be primary (complement-mediated TMA/aHUS) or secondary to endothelial injury in severe hypertension-associated TMA.6,7 This report describes biopsy-confirmed renal TMA after cocaine use presenting as hypertensive emergency, and it discusses the rationale for empiric complement blockade in the setting of persistent renal dysfunction.
Case presentation
A woman in her early 60s with a history of cocaine and tobacco use presented to the emergency department with 14 days of upper abdominal pain (initially described as right upper quadrant discomfort) and lower extremity myalgias after recent recreational cocaine use. She had no known prior diagnosis of hypertension and reported no prescription or over-the-counter medication use. She denied recent travel and had no personal or family history of autoimmune disease or sickle cell disease/trait.
On arrival, blood pressure was 209/110 mmHg. Physical examination was notable for focal epigastric tenderness without peritoneal signs. Laboratory evaluation demonstrated progressive anemia and thrombocytopenia with non-oliguric AKI. On admission, hemoglobin was 10.9 g/dL (reference 11.5–15.5), platelets 99 ×10^3/µL (reference 150–400), and serum creatinine 2.83 mg/dL (baseline 0.8 mg/dL). LDH was 908 U/L (reference 135–214), haptoglobin <10 mg/dL (reference 31–238), and high-sensitivity troponin T was 42 ng/L (reference <12). Urinalysis showed 3+ blood, 1+ protein, and granular casts (4–10 per low-power field). Urine toxicology was positive for cocaine. Echocardiography was unremarkable.
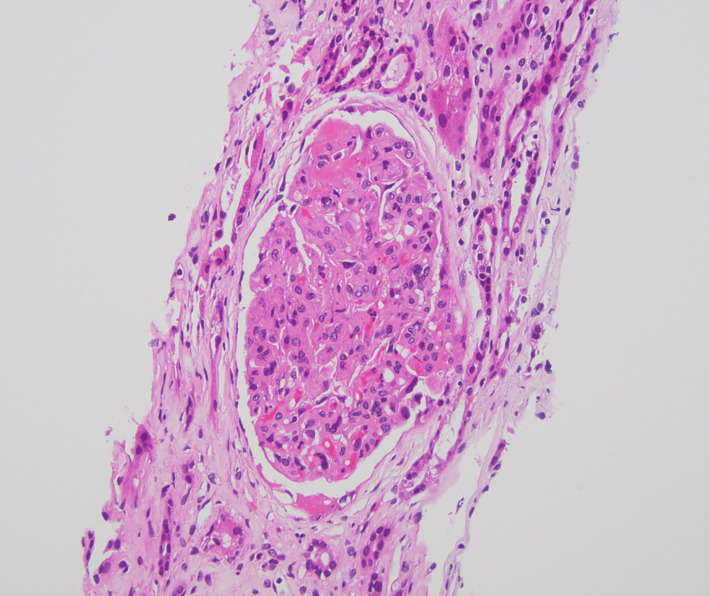
The following day, hemoglobin decreased to 8.8 g/dL and platelets to 53 ×10^3/µL. Direct antiglobulin (Coombs) testing was negative. ANA testing was negative and complement C3/C4 levels were within reference ranges. Peripheral smear demonstrated schistocytes. Hematology assessed a low likelihood of TTP given a PLASMIC score of 3 and ADAMTS13 activity of 51% (not severely reduced). Given persistent AKI with suspected TMA, the nephrology team proceeded with a right kidney biopsy. Biopsy demonstrated acute thrombotic microangiopathy involving glomeruli and small-to-medium vessels, with tubular atrophy/interstitial fibrosis and severe arterio- and arteriolosclerosis (Figure 1). Key diagnostic findings are summarized in Table 1, and relevant laboratory findings are provided in the supplementary material. Blood pressure normalized with nifedipine 60 mg daily and carvedilol 25 mg twice daily.

Despite blood pressure control, kidney function worsened during hospitalization (creatinine rose to 4.4 mg/dL). Soluble C5b-9 was elevated at 315.6 ng/mL (reference <256), indicating terminal complement activation but not establishing a primary complement-mediated etiology. Given biopsy-proven TMA and persistent renal dysfunction despite blood pressure optimization, empiric complement blockade was initiated with eculizumab after prophylactic vaccination against meningococcus serogroups A/C (MenQuadfi) and B (Bexsero) and pneumococcus (Prevnar 20). After eculizumab administration, renal function stabilized without further deterioration. The patient did not require renal replacement therapy during the index hospitalization and was discharged to continue outpatient eculizumab every 2 weeks with creatinine 4.4 mg/dL.
Six weeks after discharge, she was rehospitalized for acute heart failure with preserved ejection fraction, presenting with dyspnea, pulmonary edema, and marked lower-extremity edema. She improved with intravenous diuretics and afterload reduction and was discharged on torsemide 20 mg orally twice daily. An angiotensin receptor blocker was deferred because kidney function remained stable but had not improved. The plan was to continue eculizumab for 6–12 months with close outpatient follow-up. At three months, she remained dialysis-free with stable renal function and preserved urine output.
Discussion
This case illustrates biopsy-confirmed renal TMA presenting with hypertensive emergency after cocaine use. Importantly, cocaine exposure, malignant hypertension-associated endothelial injury, and complement activation may represent overlapping and potentially synergistic mechanisms rather than a single unifying cause. Cocaine can trigger severe hypertension and endothelial dysfunction, while malignant hypertension itself can cause TMA through shear stress–mediated endothelial injury and fibrinoid necrosis.8,9 In parallel, complement activation can be primary (complement-mediated TMA/aHUS) or secondary to endothelial injury in severe hypertension-associated TMA, and complement abnormalities have been described in hypertension-associated TMA.6,7
Distinguishing hypertension-associated TMA from complement-mediated TMA has major therapeutic implications. In hypertension-associated TMA, hematologic abnormalities often improve with blood pressure control, and any complement activation is generally viewed as secondary.9 In contrast, complement-mediated TMA is characterized by dysregulated complement activation, and organ dysfunction may persist despite hemodynamic stabilization.10,11 The Jodele diagnostic criteria for transplant-associated TMA include laboratory evidence of microangiopathy in conjunction with hypertension and proteinuria.12 Elevated soluble C5b-9 and multiorgan dysfunction were subsequently identified as high-risk features associated with clinically significant complement activation and adverse outcomes.12,13 Biomarkers of complement activation (including sC5b-9) may indicate complement involvement but are nonspecific and can be elevated in secondary TMAs.7,14 Therefore, sC5b-9 elevation should be interpreted in the clinical context rather than as definitive proof of primary complement-mediated disease. A key distinguishing feature in this case is the kidney biopsy, which confirmed acute TMA and helped rule out other intrinsic renal pathologies that could significantly change management. In patients with suspected TMA and notable kidney injury, obtaining a tissue diagnosis can meaningfully narrow the differential and enhance diagnostic certainty.
In the present case, blood pressure control stabilized hemodynamics, yet kidney function continued to worsen (creatinine increased from 0.8 mg/dL baseline to 2.83 mg/dL on admission and to 4.4 mg/dL during hospitalization). Given biopsy-proven TMA, persistent renal dysfunction despite blood pressure optimization, absence of alternative etiologies (including TTP and autoimmune disease), and evidence of terminal complement activation, the treating team initiated eculizumab empirically. Eculizumab is a humanized monoclonal antibody that binds complement protein C5, inhibiting cleavage into C5a and C5b and thereby preventing formation of the terminal complement complex C5b-9.15 Because terminal complement blockade increases susceptibility to invasive meningococcal disease, vaccination and infection-risk counseling are essential prior to treatment. In this patient, renal function stabilized after eculizumab initiation and she remained dialysis-free at three months, suggesting a complement-mediated component may have contributed to ongoing injury beyond isolated hypertension-associated TMA. It should be emphasized that the use of eculizumab in this case should be viewed as an empiric therapeutic approach for persistent organ dysfunction in a high-risk clinical setting, rather than as definitive evidence of primary complement-mediated TMA.
Conclusion
Cocaine exposure, hypertensive emergency, and complement activation can coexist in patients presenting with TMA, and these mechanisms may be overlapping rather than singular. This case highlights the importance of a thorough substance-use history and emphasizes kidney biopsy as a high-yield diagnostic tool to confirm TMA and exclude alternative renal pathologies. In selected patients with biopsy-proven TMA and persistent organ dysfunction despite blood pressure optimization, empiric complement blockade may be considered with appropriate counseling and infection prophylaxis, while acknowledging diagnostic uncertainty and the nonspecific nature of complement activation biomarkers.
Disclosures/Conflicts of Interest
None
Corresponding author
Moises Auron, MD
Department of Hospital Medicine
Cleveland Clinic
Cleveland, Ohio, USA
Email: auronm@ccf.org