Introduction
Langerhans cell histiocytosis (LCH) is a rare hematologic neoplasm characterized by aberrant clonal proliferation of mononuclear phagocytes, resulting in inflammatory and granulomatous lesions across multiple organ systems.1 While predominantly a pediatric disease with an incidence of 2-9 cases per million children,2 adult-onset LCH is significantly rarer at 1-1.5 cases per million adults.3,4
LCH involves dysregulated Langerhans cells (LC), a form of specialized dendritic cells mainly found in epidermal tissue. The pathogenesis involves MAPK pathway activation in bone marrow-derived precursor cells, leading to enhanced Langerhans cell differentiation, apoptosis resistance, and systemic inflammation.1 Clinical manifestations vary from single-organ involvement to multisystem disease, commonly including osteolytic bone lesions, diabetes insipidus from hypothalamic and pituitary lesions, pulmonary nodules, and cutaneous lesions.4 The rarity and nonspecific presentation of adult LCH contribute to diagnostic delays, with a median time from symptom onset to diagnosis reaching 8 months.5 The importance of early diagnosis lies in the devastating consequences of disease progression. If left untreated, patients with LCH can develop neurodegenerative disorders, endocrinopathies and bone-associated complications as seen in our case.1 Therefore, it is imperative that physicians understand the diagnostic criteria for LCH and include this rare disease in their differential diagnoses early to facilitate timely treatment. We present a diagnostically challenging case of adult-onset LCH with neurological manifestations, emphasizing the complexities in diagnosis and treatment.
Case Description
A 20-year-old previously healthy female presented to the emergency department with one month of intermittent sharp back pain, left-sided hearing loss, and bilateral lower extremity weakness causing gait instability. Her family history is significant for a half-sister with lupus and an unspecified type of cancer. Otherwise, she has no family history of significant neurological disease including aneurysm or subarachnoid hemorrhage. Physical examination revealed decreased strength and diminished vibration sensation in bilateral lower extremities.
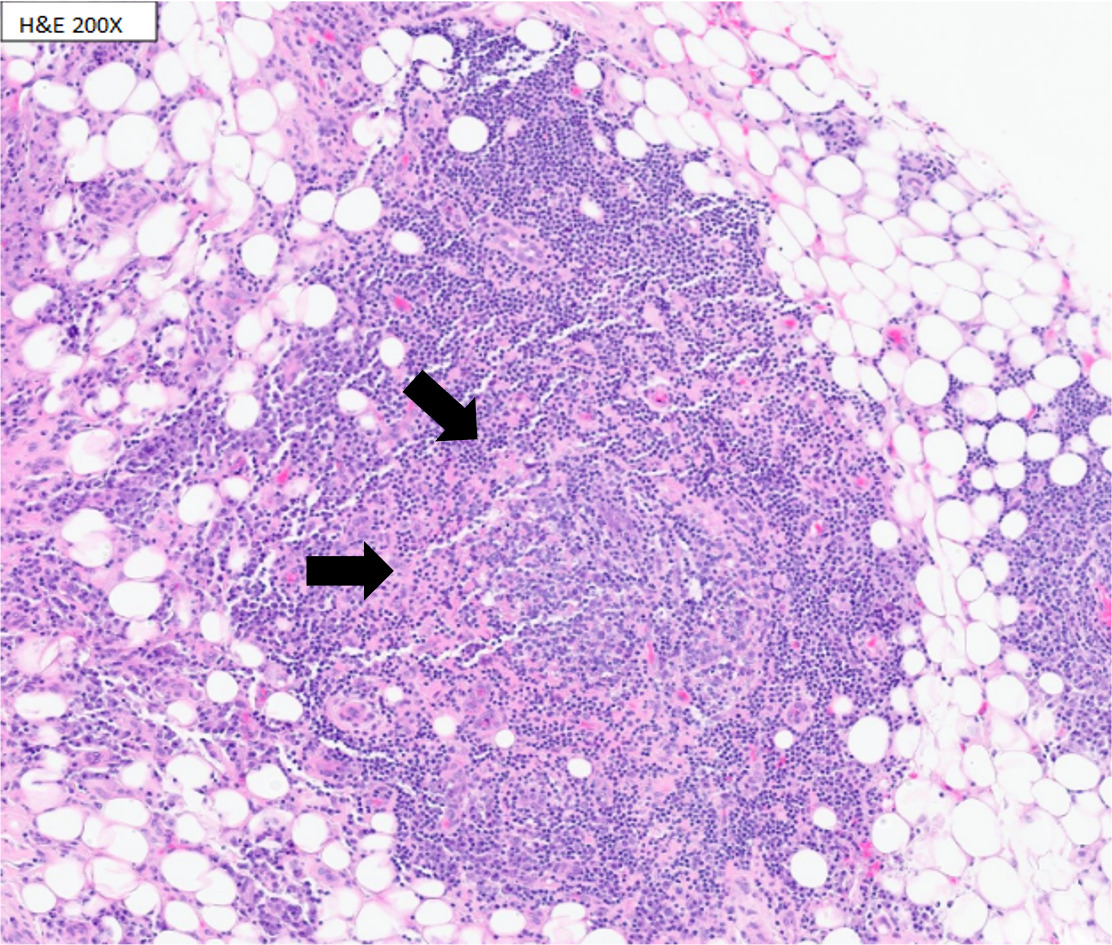
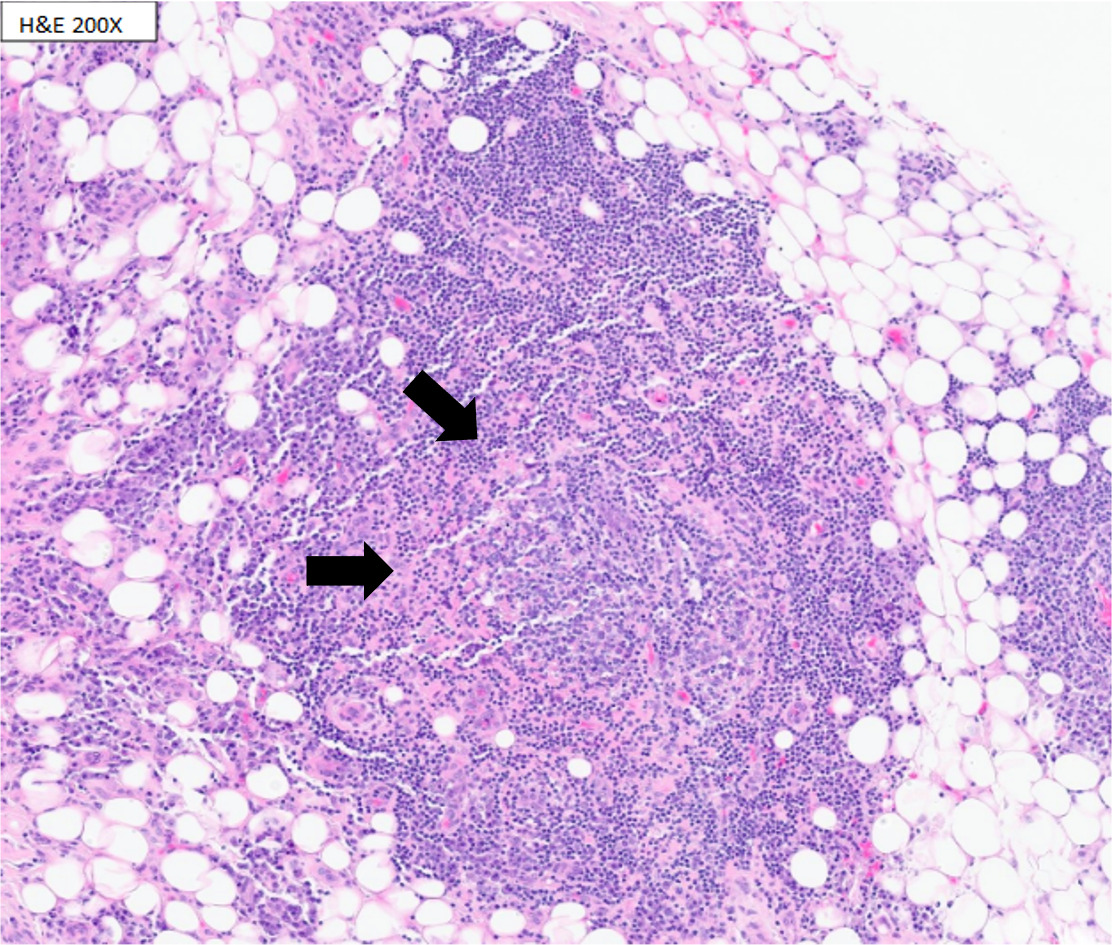
Magnetic resonance imaging (MRI) demonstrated severe spinal stenosis with cord displacement from a thoracic epidural mass at T7-9, cauda equina compression, and lumbar dural enhancement. Computed tomography (CT) scan revealed additional bilateral lucent femoral and sacral lesions. Patient underwent urgent T7-9, L3-5 laminectomy with improvement in motor function. Biopsy of the thoracic epidural mass showed atypical lymphoplasmacytic infiltration with flow cytometry revealing reactive lymphoid infiltrates but no diagnostic immunophenotype (Figure 1).
Brain MRI and Positron Emission Tomography-CT (PET-CT) for staging identified a left petrous bone lesion and mild fluorodeoxyglucose (FDG) activity consistent with postoperative changes. Subsequent petrous bone biopsy again demonstrated chronic inflammatory infiltrates with lymphocytes, plasma cells, and histiocytes without a definitive diagnosis. Extensive infectious and rheumatologic workups were negative, including syphilis, tick-borne illnesses, malaria, tuberculosis, and varicella. Despite an elevated serum IgG4 level, there were no clinical signs consistent with IgG4-related disease. During this hospitalization, multiple consult services were involved including neurology, neurosurgery, otolaryngology, pediatric hematology, hematology, orthopedics, rheumatology and pediatric rheumatology. Given her complicated clinical picture, the list of working diagnoses at the time included non-Hodgkin’s lymphoma, IgG4-related disease and chronic recurrent multifocal osteomyelitis (CRMO). However, her presentation, pathological evaluation and laboratory evidence did not fully support any of these diagnoses. On hospital day 19, the patient developed acute lower extremity weakness. Urgent MRI showed possible thoracic spinal edema or myelomalacia, which improved with high-dose corticosteroids. Patient was then discharged to a rehabilitation facility on hospital day 54 with a steroid taper.
Two weeks after discharge, the patient was readmitted with dysphagia and left tongue deviation. Cervical MRI revealed a mass abutting cranial nerve XII with mastoiditis and cranial nerves IX-X dural enhancement. She underwent methylcellulose injection for left vocal cord augmentation. Bone marrow biopsy showed normal trilineage maturation with no neoplastic process. Peripheral blood and bone marrow flow cytometry revealed polytypic T cells, polyclonal B cells, a normal blast count, and a normal karyotype. Given the patient’s complicated clinical picture, response to glucocorticoids, and initial biopsy demonstrating atypical lymphoplasmacytic infiltration, a hematologic neoplasm was suspected, most notably non-Hodgkin’s lymphoma. However, the biopsy findings were not definitively consistent with lymphoma, and the overall picture was more suggestive of a broader malignant hematological process yet to be identified. IgG4-related disease and CRMO were also considered but were not supported by the clinical presentation, pathological evaluation, and laboratory evidence. Therefore, repeat outpatient PET-CT and biopsy were planned for outpatient evaluation.
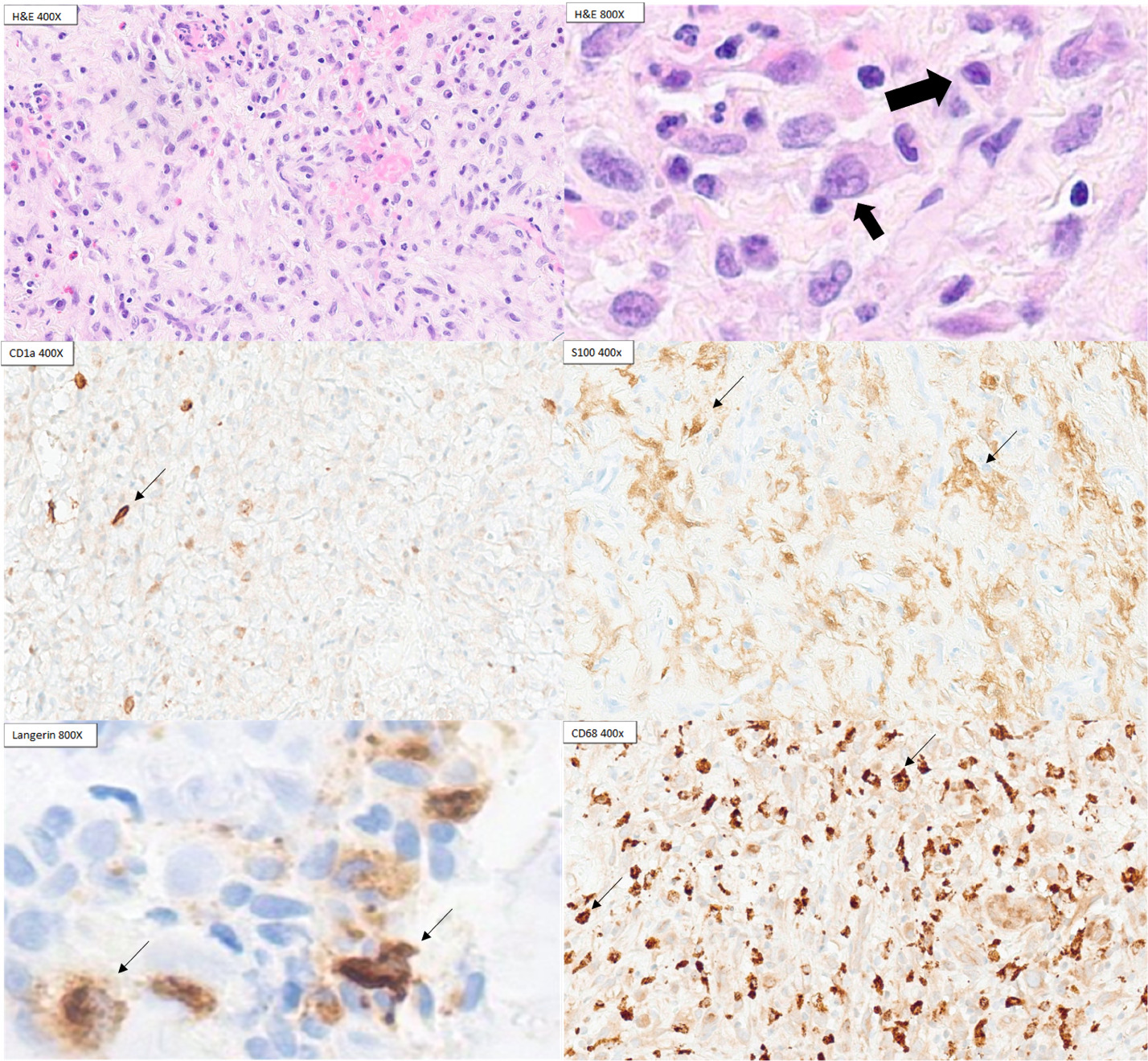
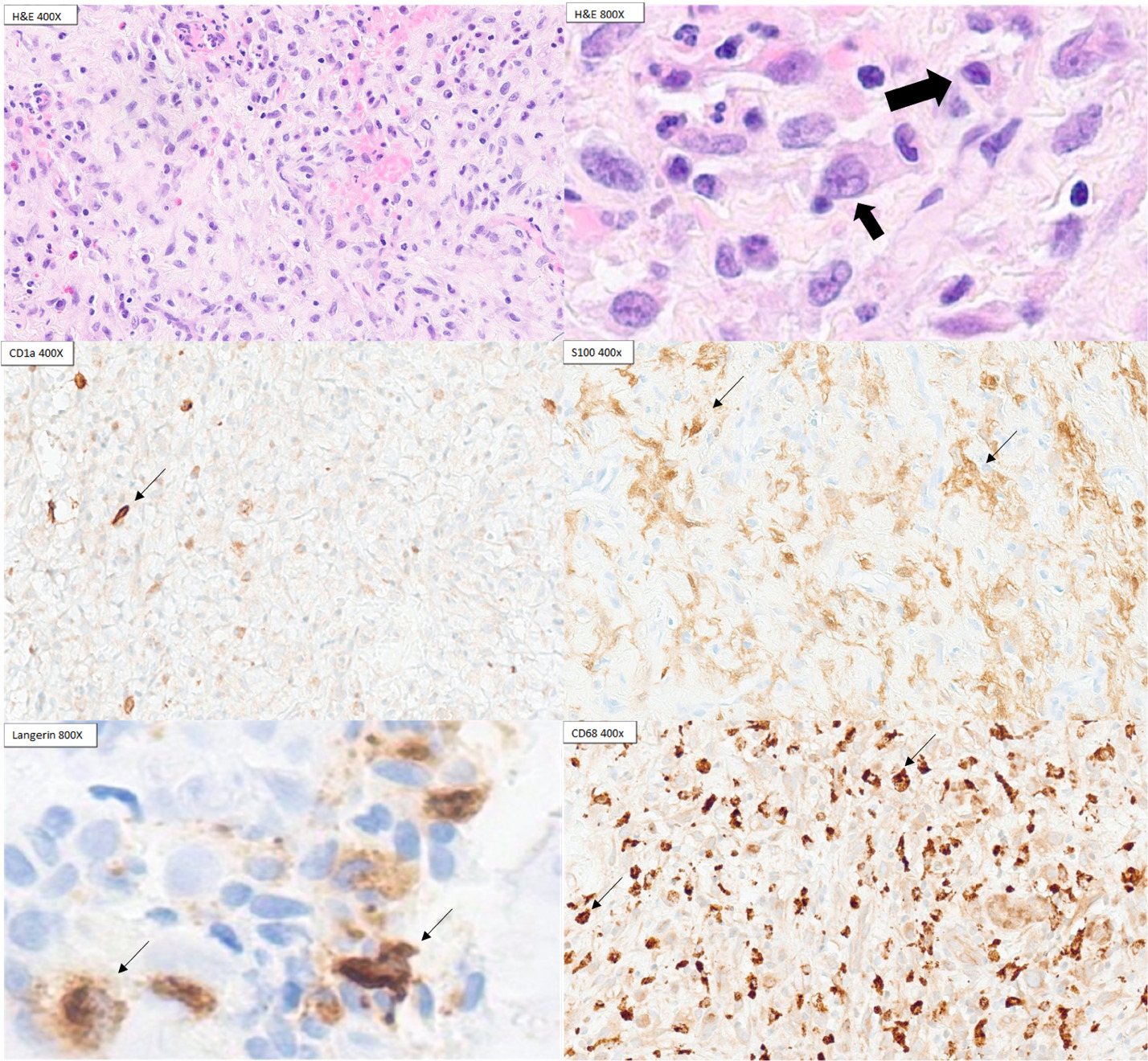
Six weeks after her second admission, the patient returned with blurry vision and left lateral gaze palsy. Repeat MRI showed interval enlargement of the left petrous mass with likely cranial nerve VI compression. A third petrous apex biopsy finally revealed inflammatory infiltrates with histiocytes positive for CD68 and a subset of cells positive for CD1a, S100, and Langerin, establishing the diagnosis of LCH. BRAF V600E immunohistochemistry was negative (Figure 2).
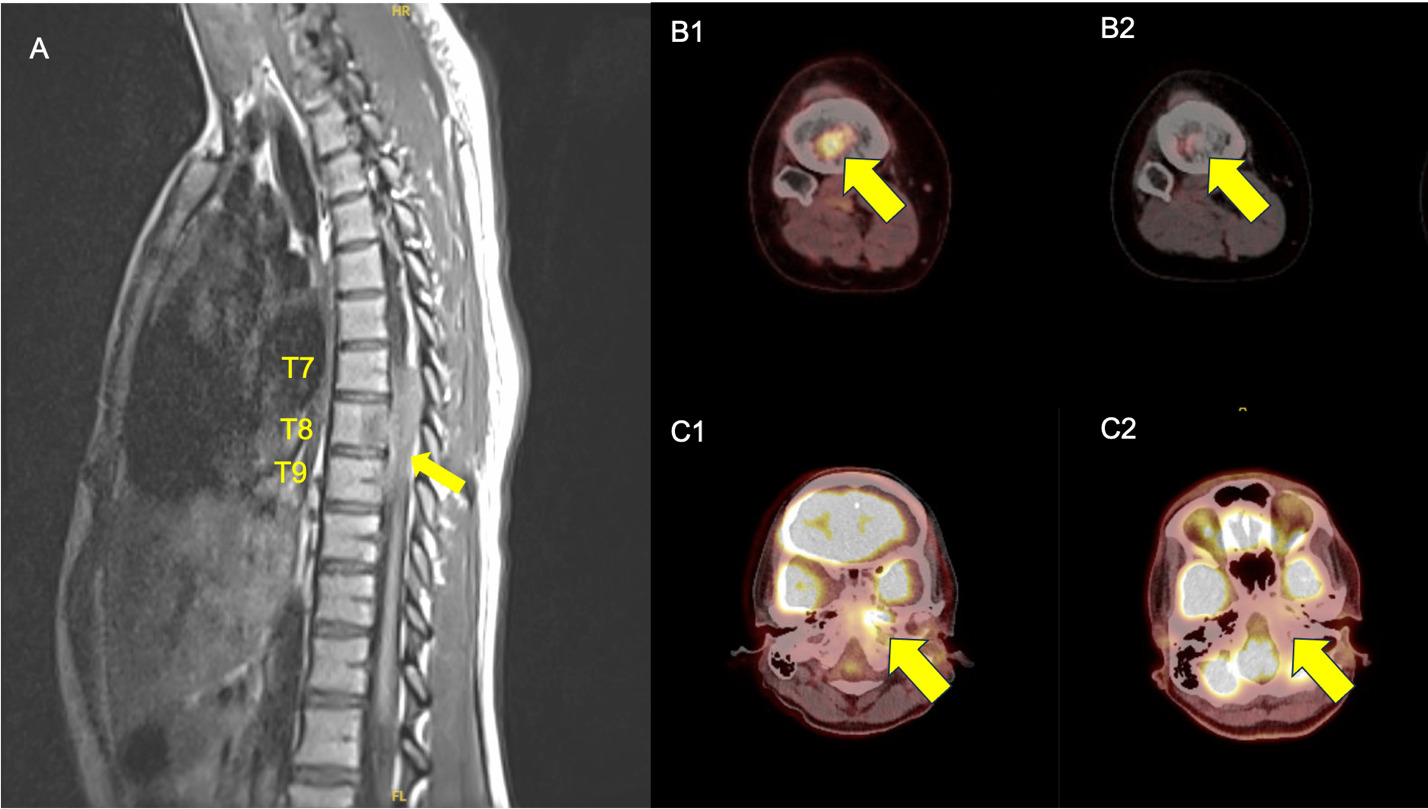
Initial treatment with zoledronic acid 4 mg per cycle failed, with disease progression after two cycles. Treatment escalated to cytarabine 150 mg/m² monthly, but PET-CT after two cycles demonstrated disease progression in the left mastoid and a new right proximal tibia lesion. Therapy was switched to cladribine 0.14 mg/kg daily with hydroxyurea. Treatment was complicated by Elizabethkingia meningoseptica-associated central line infection during cycle 2, attributed to the patient’s pet lizard. The patient was hospitalized and treated with antibiotic therapy. PET-CT after cycle 2 of cladribine and hydroxyurea showed decreased left petrous, mastoid, and right tibial lesions, though increased FDG activity appeared in the left lateral pelvis (Figure 3). Treatment continued with intermittent hydroxyurea cessation due to neutropenia. By cycle 6, the patient’s diplopia resolved, and imaging showed continued improvement with complete resolution of the tibial lesion. During cycle 8, the patient developed bilateral hip avascular necrosis which was managed conservatively. After 9 cycles, the most recent PET-CT during cycle 8 showed no evidence of active disease.
_t1-w.png)
Discussion
This case of bone-associated multifocal, single-system LCH exemplifies the diagnostic and therapeutic challenges of adult LCH. LCH is extremely rare with an estimated incidence of 2-9 cases per million patients in children and 1-1.5 cases per million patients in adults, which contributes to the diagnostic challenge.3,4 The diagnosis of LCH largely relies on pathologic evaluation of tissue biopsy in addition to mutation markers but a clinical diagnosis can be made in characteristic cases when there is atypical or insufficient pathologic evidence.4 Some characteristic clinical features of LCH include upper lobe nodular/cystic lung lesions, central diabetes insipidus, and punched-out lytic bone lesions typically involving flat bones.4 If patients have undergone FDG-PET, the site with greatest FDG activity should be biopsied if accessible and multiple cores should be obtained to maximize diagnostic likelihood.4
During pathologic evaluation, LCH typically expresses benign LC markers including CD68, S100, CD1a and Langerin, making it challenging to distinguish LCH from reactive LC proliferation.4 LCH can be differentiated from reactive LCs by a more diffuse CD1 expression and BRAF-V600E mutation analysis.4 Furthermore, the diagnosis of LCH can be difficult to achieve given the presence of numerous mimickers such as IgG-4 related disease, Erdheim-Chester Disease (ECD) and Rosai-Dorfman Disease (RDD).6 ECD and RDD are clinically similar to LCH and require histologic evaluation These two disorders may show similar histiocyte markers on immunohistochemistry and may require prudent pathologic evaluation to differentiate from LCH.6 In addition, the possibility of histiocytic sarcoma was considered but not supported by pathological evidence given the lack of CD 163, lysozyme with the presence of CD1a and Langerin.7 Reactive Langerhans cells (LC) can be from many other diseases such as infections, rheumatologic disorders such as systemic lupus erythematosus and lymphoma. However, the presence of such conditions was evaluated by the workup previously listed above. Conditions without reactive LCs can also be confused with LCH, such as fibroinflammatory disorders including IgG4-related disease. In our patient’s case, the patient was younger than the typical population who has IgG4-related disease.8 In addition, she does not have classic presentations such as glandular tissue involvement but instead has lytic bone lesions.8 Furthermore, her pathology is not consistent with IgG4-related disease, with the absence of storiform fibrosis and obliterative phlebitis.8 Treatment of bone-only LCH starts with bisphosphonate, methotrexate or hydroxyurea whereas the mainstay of multisystem LCH treatment is vinblastine with prednisone or antimetabolite-based treatments such as cytarabine and cladribine.4,9
In our patient’s case, spinal epidural masses and cranial neuropathies are rare LCH manifestations, contributing to diagnostic complexity and omission from differential diagnoses.10–12 For example, IgG4-related disease can present with neurologic deficits which can be similar to those manifested in this patient.8 The nonspecific histologic findings and sparse neoplastic cells in LCH tissue samples often necessitate multiple biopsies with appropriate immunohistochemical staining for CD1a, S100, and Langerin.4 In fact, informing the pathologist of the likelihood of LCH is often required to establish the diagnosis, given that histologic findings often overlap with fibroinflammatory diseases, making the differentiation between alternative diagnoses extremely difficult.13 As demonstrated in our patient, three biopsies were required before definitive diagnosis, during which cranial nerve VI palsy developed.
The initial steroid response suggested a steroid-responsive malignancy such as lymphoma. This emphasizes the importance of tissue diagnosis. Bone marrow evaluation may be necessary to exclude concurrent hematologic neoplasms, which can coexist with adult LCH. Treatment selection remains challenging due to the absence of adult LCH clinical trial data, with most regimens extrapolated from pediatric studies.1 For unifocal disease, local treatment such as surgery is preferred,4 while multisystem disease typically requires systemic therapy. Bisphosphonates are first-line treatment for single-system polyostotic disease due to their favorable toxicity profiles, acting by inhibiting osteoclast activity and reducing cytokine release by LCs, thereby reducing bone lysis and pain.4,9 When bisphosphonates fail, as in our patient, the recommended treatment for adult multisystem neurodegenerative LCH mainly involves empirically derived chemotherapy, specifically cytarabine, cladribine, and hydroxyurea.1 Systemic therapies carry significant hematologic toxicity risks, including severe immunosuppression and infection.14 Pediatric studies report universal WHO Grade 4 hematologic toxicity with cytarabine-cladribine combination therapy, with documented severe infections in all patients.14 Our patient experienced similar toxicity with treatment-related sepsis, highlighting these therapies’ risks.
Novel and alternative therapies are currently being studied. Emerging targeted therapies, including BRAF V600E inhibitors like vemurafenib, show promise but demonstrate only 43% response rates in adults with frequent relapses.15 Alternative agents such as clofarabine [NCT02425904] are under investigation. The challenge remains developing safer, more effective treatments specifically for adult patients.
Conclusion
Adult-onset LCH presents significant diagnostic and therapeutic challenges due to its rarity, variable presentation, and limited treatment data. In addition, LCH therapy in adults lacks high-quality data from clinical trials, which is much needed given devastating symptoms as well as poor prognosis. Clinicians should maintain high suspicion for LCH in atypical presentations, particularly with neurological involvement, and aggressively pursue tissue diagnosis to avoid misdiagnosis and delayed diagnosis. Current systemic therapies, while potentially effective, carry substantial toxicity risks, underscoring the urgent need for adult-specific clinical trials and safer treatment options.
Disclosures/Conflicts of Interest
The authors have no conflicts of interest to disclose.
Corresponding author
Zhan Rong M.D.
Department of Internal Medicine
Stony Brook University Hospital, NY, USA
Email: zhan.rong@stonybrookmedicine.edu