
Case Presentation
A 19-year-old woman presented to hospital complaining of fever, rash, and neck swelling. She reported experiencing fatigue and malaise approximately two weeks prior to presentation. She reported neck swelling at the same time, most prominent on the left side. She went to the student clinic of her university where she was tested for mononucleosis and strep throat which were negative.
Several days prior to presentation, she began to experience daily fevers, most prominent in the evening. Over the prior two days she developed a skin rash, which began on her arms and spread to the rest of the body. On the morning of presentation, she was noted to have a fever of 103° Fahrenheit which alarmed her and prompted her to come to the hospital emergency department for further evaluation.
She also reports associated poor appetite, weight loss but denies any odynophagia, dysphagia, chest pain, palpitations, shortness of breath, cough, nausea, vomiting, abdominal pain, lower extremity swelling, dysuria, loss of consciousness or joint swelling.
Her medical history included beta-thalassemia trait. She took no medications. She had no previous surgeries and no known medication allergies. She did not use tobacco products, drink alcohol and denied recreational or other illicit drug use. She was an international student at a local university and was born in Thailand. Her maternal grandfather suffered from lymphoma.
On physical examination her temperature was 103 °F, pulse rate 89 beats per minute, respiratory rate 16 per minute, blood pressure 93/52 mmHg, and pulse oxygen saturation of 98% on ambient air. She was alert and awake, oriented to person, place and time, and not in acute distress. Bilateral cervical lymphadenopathy was found, most prominent on the left side. A 1mm ulcer at the apex of the frenulum linguae was noted. Cardiovascular and pulmonary examinations were unremarkable. Abdomen was soft and scaphoid, non-tender to palpation, without organomegaly. Extremities were without cyanosis, clubbing or edema. No flank or suprapubic tenderness was noted. The patient demonstrated intact higher mental functioning, cranial nerves I-XII were intact, and muscular strength was preserved throughout. Mixed blanching, erythematous papular/patchy rash noted on chest, abdomen, extremities, sparing the palms and soles.
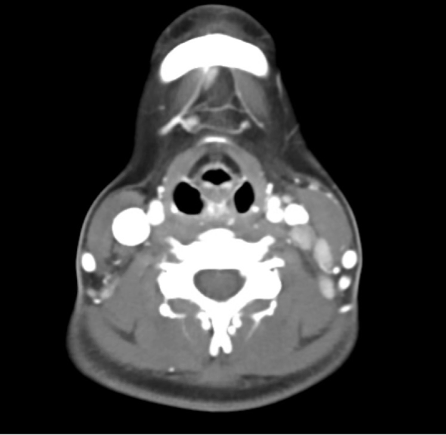
See Table for relevant laboratory data. See Figure 1 for relevant radiographic studies.
_with_evidence_of_left_sided_cervical_lymphadeno.png)
Warren Alpert Medical School Student Presentations
Student (The Miriam Hospital)
Working Diagnosis: Systemic Lupus Erythematosus (SLE)
This patient’s fatigue, fevers, myalgia, and weight loss with decreased appetite are consistent with the presentation of SLE. Although the most common skin manifestation of SLE is a facial eruption, skin involvement in SLE is highly variable, and could absolutely explain this patient’s erythematous, patchy, papular rash. Oral ulcers are also common in SLE. Leukopenia and lymphadenopathy, both present in this patient, each occur in about 50% of patients with SLE, with lymph node swelling occurring most often at initial disease presentation, as would be the case here. An autoimmune condition like SLE is also in line with this patient’s two-week presentation with gradual onset of symptoms. I recommend serological testing with anti-nuclear antigen (ANA) and anti-double stranded DNA antibodies.
Student (Rhode Island Hospital)
Working Diagnosis: Adult Onset Still’s Disease (AOSD)
A rare clinical diagnosis of exclusion, the hallmarks of AOSD include quotidian fever and a mixed erythematous or salmon-colored maculopapular, patchy rash that covers the extremities and trunk. Lymphadenopathy, particularly cervical lymphadenopathy, is common. Lymph nodes in AOSD are often avidly enhancing, as were those of our patient. C-reactive protein is universally elevated. Myalgias and arthralgias are also extremely common, whereas arthritis is not. Anorexia and weight loss are also common. However, laboratory evaluation of AOSD often reveals leukocytosis, whereas our patient presents with leukopenia. AOSD will often present with negative ANA and RF labs, if obtained. AOSD commonly presents with pharyngitis, which our patient does not report. If other conditions are ruled out, a clinical scoring scale such as the Yamaguchi criteria could be utilized to diagnose AOSD in this patient.1
Student (Kent County Hospital)
Working Diagnosis: Human Immunodeficiency Virus (HIV) infection
This patient’s relatively nonspecific subjective symptoms, including fevers, chills, weight loss, rash, myalgia/arthralgia, headache with unknown drug use or sexual history could be consistent with acute onset HIV infection. It seems like her beta thalassemia trait status is relatively mild, but it is possible that she may have needed blood transfusions in the past, which may have led to HIV inoculation. Acute HIV infection occurs in the first 6-12 weeks after infection with viral replication occurring in lymphoid tissues. Acute HIV infection is generally marked by non-specific protean symptoms of fever, chills, malaise, rash and lymphadenopathy consistent with our patient’s presentation. Serum HIV RNA viral load testing would be the next step in diagnosis.
Student (Veterans Affairs Medical Center, Providence, RI)
Working Diagnosis: Kikuchi-Fujimoto Disease (KFD)
KFD, which is a benign, self-limited syndrome characterized by lymphadenopathy (often unilateral in the cervical region), fever/night sweats, and rash/skin manifestations- the three presenting symptoms from our patient. Other clinical features of KFD include weight loss, myalgias, fatigue, and arthritis. It is extremely rare worldwide but has a higher prevalence in young women under the age of 30. The onset of KFD is often subacute with symptoms evolving over 2-4 weeks and usually self-resolving within 4 months. It is estimated that 40% of patients with KFD will have cutaneous manifestations which include morbilliform rashes, indurations, maculopapular rashes, and oropharyngeal ulceration; the latter two of which was seen in our patient. The classic lab finding in KFD is leukopenia. KFD is highest on my differential given the alignment with the patient’s case (timeline, regional lymphadenopathy, fevers, rash, ulcer, leukopenia, elevated CRP, and CT findings). Diagnosis is confirmed via lymph node biopsy.
Dr Joseph Garland
The initial differential diagnosis for this patient is quite broad. The short duration of symptoms with a constellation of fever, rash, fatigue, malaise, and prominent cervical lymphadenopathy, and subsequent evolution of a maculopapular rash, strongly suggest acute infectious etiologies. Initial presentation of rheumatologic conditions or malignancy are rarer but also possible etiologies. Perhaps the most differentiating of these nonspecific symptoms is the prominent lymphadenopathy. Her imaging notes the presence of axillary lymphadenopathy, which is points to a systemic process rather than one localized to the cervical chain. A sexual history was not presented, which would be helpful history to have, as would a thorough lymph node exam.
My differential would begin with infectious etiologies. Acute viral disease, particularly acute HIV disease, would be most likely. A mononucleosis syndrome due to EBV or CMV is also possible, though rash is not common. Among bacterial infections, syphilis and Group A Streptococcus would seem the most likely possibilities. And among other kingdoms, I would consider toxoplasmosis, though she has had no exposures. For non-infectious etiologies, acute lymphoma and lymphadenopathy syndromes including Kikuchi-Fujimoto Disease and Castleman Disease are possible but rare, and the presentation has aspects that are not typical for each. Further, they involve a more invasive diagnostic approach, so I would begin with evaluating the more likely infectious etiologies with simple and efficient testing. I would start with HIV testing with a 4th-generation antibody/antigen test.
Hospital Course
The patient was admitted to hospital. Serological assays for HIV and SLE were non-reactive. An excisional lymph node biopsy was performed.
Dr Habibe Kurt
The review of the sections showed an enlarged lymph node (1.8 cm in greatest dimension) with multiple, pale circumscribed foci in the paracortical areas (Figure 2A). Focal areas of dermatopathic changes, paracortical hyperplasia, and rare reactive germinal centers were also noted. Those pale areas are composed of numerous histiocytes, lymphocytes, and apoptotic bodies (Figure 2B). A subset of histiocytes showed crescent-shaped nuclei (Figure 2B, arrows). However, there were no neutrophils in the necrotic areas, which are usually associated with infectious diseases. There were also no large, highly atypical cells with prominent nucleoli [Reed Stenberg (RS)-like cells], which are seen in classic Hodgkin’s lymphoma. Immature cells with blastoid morphology were also not noted, predominantly excluding lymphoblastic lymphoma. The immunohistochemical stains were performed to further rule out lymphoma. The necrotic areas were predominantly composed of CD68 and MPO positive histiocytes (Figure 2C&D) and CD3 positive T-cells (Figure 2E) with scattered CD20 positive B-cells (Figure 2F). The absence of CD20 positive B-cell proliferation excludes B-cell lymphomas, especially Burkitt lymphoma commonly seen in this age group. CD8 positive cytotoxic T-cells (Figure 2G) predominated in these areas compared to CD4 positive T-helper cells (Figure 2H). CD123 stain highlighted increased plasmacytoid dendritic cells around the necrotic areas. Occasional immunoblasts were seen by CD30 stain; however, no RS-like cells were seen. Although there was no significant plasma cell proliferation on H&E stained sections, occasional plasma cells were highlighted by CD138 stain. Flow cytometry immunophenotypic studies performed on a portion of the specimen identified no aberrant B- or T-cell proliferation. CD4:CD8 ratio was 1.2:1, as observed in the tissue section.

In summary, pale subcortical areas mainly composed of MPO-positive histiocytes with crescent-shaped nuclei, CD8+ T-cells, increased plasmacytoid dendritic cells, and numerous apoptotic bodies without neutrophils and significant plasma cell proliferation are diagnostic for KFD. The main differential diagnosis is SLE, and correlation with serologic studies is essential to rule out this disease since differentiation of these two entities solely on morphologic evaluation is difficult. The morphologic findings in acute HIV lymphadenitis usually include florid follicular hyperplasia, and the findings described above are not seen.
Final Diagnosis
Kikuchi-Fujimoto Disease
Discussion
Kikuchi-Fujimoto disease, or histiocytic necrotizing lymphadenitis, is characterized by tender lymphadenopathy, fever and other systemic symptoms.2 Young adults (age <40years) are commonly affected, and although first reported in Japan, the condition has since been reported worldwide in a variety of ethnic backgrounds. KFD is a self-limited inflammatory illness without an identified cause though viral infection (Parvovirus B19, Ebstein-Barr virus and others) and autoimmune etiologies are thought to be the most likely explanations.3 The clinical presentation is non-specific but findings of regional lymphadenopathy and fever, in the absence of other causes should prompt consideration of KFD.4 Systemic lupus erythematosus, lymphoma and human immunodeficiency virus infection should be considered in the differential. Diagnosis is made via histopathology which reveals patchy areas of necrosis and histiocytes at the edge of necrotic areas. The disease course is self-limited, and symptoms may resolve within several months of onset, with a low recurrence rate of 3-4%.5
Acknowledgements
Brandon Kelley, Rex Nathaniel, Dominique Dockery, Winston McCormick
Conflicts of Interest
The authors declare they have no conflicts of interest.
Corresponding author
Arkadiy Finn, MD
Assistant Professor of Medicine, Clinician Educator
Warren Alpert Medical School at Brown University
Division of Hospital Medicine
The Miriam Hospital, 164 Summit Avenue, Providence, RI 02906