Background
It is clinically difficult to differentiate thrombotic thrombocytopenic purpura (TTP) from other causes of thrombotic microangiopathy (TMA) such as hemolytic-uremic syndrome, atypical hemolytic-uremic syndrome and drug-induced thrombotic microangiopathy. Infection, malignant hypertension, pregnancy, drug reactions and malignancy may present with similar features. The uniting feature of these presentations is the presence of microangiopathic hemolytic anemia (MAHA) and thrombocytopenia with or without renal dysfunction, fever, neurological deficit or variable pattern of systemic tissue injury from microvascular thrombosis. This is usually referred to as the classical “pentad” of TTP.1
Case Presentation
A 28-year-old woman with congenital human immunodeficiency virus (HIV) infection and hypertension, presented to hospital with dry cough, watery diarrhea, abdominal pain with nausea and vomiting. Past history was significant for poorly controlled HIV infection and previous hospitalization for pre-eclampsia and intrauterine fetal demise five months before this presentation. Blood pressure at presentation was 190/120 mmHg. Physical exam showed a thin built woman with icteric sclera with no evidence of cutaneous bleeding. Blood analysis showed elevated creatinine 2.2 mg/dl from her baseline of 1.6 mg/dl. Her platelet count was far below her baseline of 68,000 cells/ml at 10,000 cells/ml.
Evidence of intravascular hemolysis was present with 3 schistocytes per high power field, low serum haptoglobin less than 10 mg/dl, and elevated serum lactate dehydrogenase (LDH) above 1600 IU/L. Her hemoglobin was 11 g/dL. Her initial PLASMIC score was calculated at 5 for thrombocytopenia less than 30,000 cells/ml, hemolysis, lack of cancer history and transplantation, and international normalized ratio (INR) less than 1.5. Disseminated intravascular hemolysis (DIC) was thought unlikely given elevated fibrinogen above 600 g/L with normal prothrombin (PT) and partial thromboplastin times (PTT). Unfortunately, the patient left the hospital emergency department against medical advice.
Three days later, she was found by her partner unresponsive and returned to hospital. She was nonverbal but able to follow commands. Computed tomography (CT) of the head was negative for hemorrhage. Labs showed progressive decline in platelets to 5000 cell/ml with worsening hemolysis. Hemoglobin declined to 8 g/dL. Worsening renal dysfunction was present with serum creatinine 2.5 mg/dl. Schistocytes on blood smear were re-demonstrated. Her CD4 T cell count was only 26 cells/mm3. Her blood pressure was 210/150 mmHg and she was afebrile. Chest x-ray was unremarkable and mild leukocytosis was evident at 12,000 cells/ml. A presumptive diagnosis of TTP was made; the patient underwent emergency plasmapheresis and was initiated on intravenous high dose corticosteroids. She required mechanical ventilation and nicardipine infusion to control hypertension. She developed a generalized tonic-clonic seizure and, shortly thereafter, cardiac arrest. A broad workup for neurological deterioration was negative for bacterial, viral and opportunistic infections of the central nervous system including encephalitis/meningitis PCR panel, VDRL and JC virus were nonreactive.
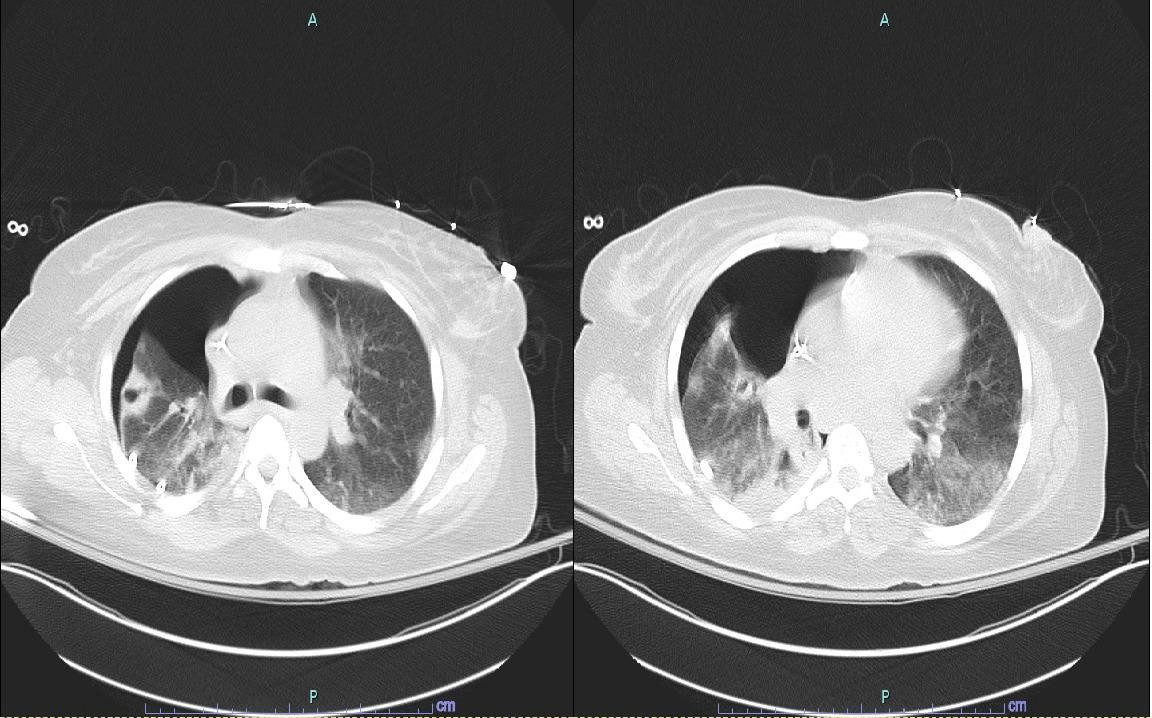
A repeat chest x-ray showed new bilateral basal opacities, white blood cell count (WBC) was gradually increasing with left shift. After finishing 5 sessions of plasmapheresis, TTP was believed to be unlikely given ADAMTS13 activity of greater than 100 % and persistent MAHA and thrombocytopenia. Blood cultures grew cefepime-sensitive Pseudomonas aeruginosa, and antibiotic coverage was initiated with cefepime. The addition of an aminoglycoside was considered but not implemented considering worsening renal function and need for intermittent hemodialysis. Her leukocytosis and left shift failed to improve. She developed a unilateral pneumothorax requiring chest tube placement. CT of the chest showed bilateral but predominantly right sided apical cavitary lesions (Figure 1). Histoplasma urine antigen and cryptococcal antigens were non-reactive. Negative methenamine silver and acid-fast bacilli staining of bronchoalveolar lavage (BAL) samples made Pneumocystis jirovecii and Nocardia spp. infection less likely.
_of_the_chest_showing_bilateral_cavitary_pulmonary_lesions.jpeg)
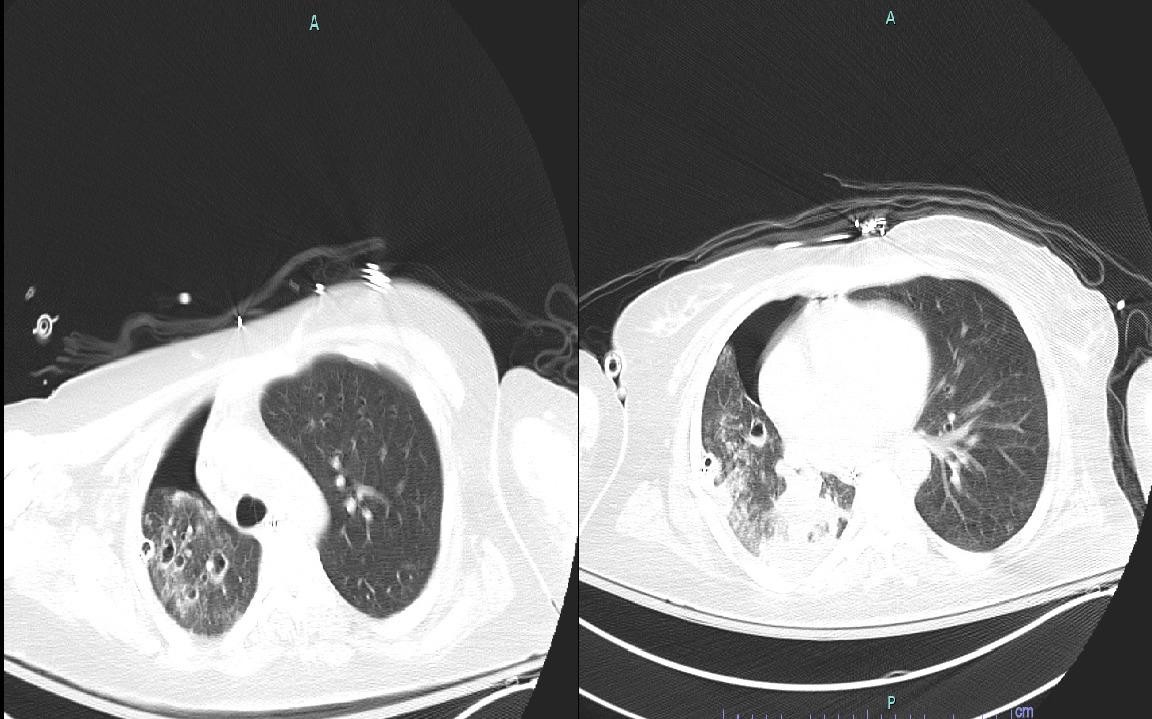
Evidence of continuous active infection despite cefepime monotherapy prompted repeat workup. A repeat CT chest revealed progression of cavitary lesions (Figure 2). BAL sample bacterial culture grew a cefepime-resistant P. aeruginosa. Antibiotic coverage was changed to meropenem and tobramycin. Findings of infection including leukocytosis, left shifting of the differential, as well as disappearance of schistocytes with stabilization of the red cell and platelet counts.

Ultimately the patient did not demonstrate neurological recovery, leading to a decision to withdraw curative measures and establish comfort care.
Discussion
Systemic infections may mimic the presenting clinical features of TTP. In the Oklahoma TTP-HUS (hemolytic-uremic syndrome) Registry, 1989–2010, 7% of the presenting clinical features were subsequently attributed to a systemic infection.1 Infections with 41 different bacteria, viruses, and fungi were documented, suggesting that many different systemic infections may mimic the presenting clinical features of TTP.2 Only one case of Pseudomonas infection in the registry was reported. A common clinical belief is that if more features of the “pentad” are present and if the abnormalities are more severe, then the diagnosis of TTP is more likely. However, the “pentad” is relatively rare among patients with TTP with no alternative disorders and in whom the diagnosis is supported by the presence of severe ADAMTS13 deficiency.2 Clinicians should remain open to other diagnoses even when the complete TTP “pentad” of clinical features is present, particularly in the presence of fever and chills indicating possible infection.
Pathophysiology of TTP involves deficiency of ADAMTS13, which is a metalloprotease enzyme responsible for breakdown of the large Von Willebrand factor (VWF). Such deficiency, in the presence of a trigger like pregnancy, pancreatitis or infection, can lead to accumulation of VWF, endothelial injury, platelet aggregation and ultimately widespread microthrombi formation.3 The presence of severe ADAMTS13 deficiency supports the clinical diagnosis of TTP but ADAMTS13 activity values in isolation neither establish nor exclude the diagnosis of TTP. In the management of patients with suspected TTP, the dominant criterion for initiating or discontinuing plasmapheresis should be the absence or presence of an alternative etiology for the microangiopathic hemolytic anemia and thrombocytopenia.4
PLASMIC score is a recently developed clinical tool to predict low ADAMTS13 activity and hence TTP probability. A recent retrospective Canadian study showed that a high-risk (6-7) PLASMIC score successfully predicted patients with severe ADAMTS13 deficiency with a sensitivity of 96.7% but low specificity of 30.7%. The positive predictive value of the PLASMIC score is 45.7% while the negative predictive value is 93.9%, making it a useful supplement but not a replacement for clinical judgment.5
P. aeruginosa should be suspected in cases rapidly progressive cavitary pneumonia. Although any lung region can be involved, the right upper lobe is the most frequently affected.6 P. aeruginosa is rarely identified as the pathogenic agent in community-acquired pneumonia, accounting for only 0.4–6.9% in reported cases of CAP requiring hospitalization and 1.8–8.3% in CAP requiring ICU admission.7 Risk factors for Pseudomonas pneumonia include structural lung disease, recurrent exacerbations of COPD requiring corticosteroid/antibiotic treatment, antibiotic use, immunocompromised status, chronic heart failure, cerebrovascular disease, advanced age and smoking. In severe P. aeruginosa pneumonia, mortality of patients who develop progressive septic shock and multiple organ dysfunction (MOD) can reach 50–100%.8,9
If Pseudomonas pneumonia is suspected in critically ill patients admitted to the ICU, guidelines recommend use of an antipseudomonal β-lactam (piperacillin-tazobactam, cefepime, imipenem, or meropenem) plus an antipseudomonal fluoroquinolone; or the above β-lactam plus an aminoglycoside and azithromycin; or the aboveβ-lactam plus an aminoglycoside and a fluoroquinolone.10 Once P. aeruginosa is confirmed to be the pathogenic agent, the treatment regimen should be adjusted to be more targeted. Targeted therapy recommended by guidelines includes an antipseudomonal β-lactam plus an aminoglycoside or a fluoroquinolone, with the alternative being an aminoglycoside plus a fluoroquinolone. As this case demonstrates, the importance of dual coverage of pseudomonas infection is of paramount importance as it may acquire resistance rapidly during treatment.
Author Contribution
All Authors (MAA, OF, MG, DG) have reviewed the final manuscript prior to submission. All the authors have contributed significantly to the manuscript, per the ICJME criteria of authorship.
-
Substantial contributions to the conception or design of the work; or the acquisition, analysis, or interpretation of data for the work; AND
-
Drafting the work or revising it critically for important intellectual content; AND
-
Final approval of the version to be published; AND
-
Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Disclosure
Authors have nothing to disclose.
Funding
No funding to disclose.
Corresponding Author
Mohammad Abu-Abaa, MD
Capital Health Regional Medical Center Internal Medicine Residency Program 750 Brunswick Ave, Trenton, NJ
Email: Mabu-abaa@capitalhealth.org
Phone: (609) 394-6000
ORCID ID 0000-0003-1752-1235