BACKGROUND
Most antibiotics lead to important alterations of intestinal microbiota, often resulting in diarrhea. Antibiotic associated diarrhea (AAD) is defined as passage of three or more unformed stools per day beginning anywhere from within a few hours up to 8 weeks after starting antibiotic therapy.1,2 The frequency of AAD has been reported to occur in 5-35% persons exposed to antimicrobials and varies upon the class of antibiotics received, health of the host and exposure to pathogens.3 AAD may range from mild, self-limiting illness to fulminant, pseudomembranous colitis that can be life threatening.4,5
The first cases of antibiotic-associated colitis were described in 1950s and 1960s soon after the introduction of antimicrobial drugs with activity against Staphylococcus aureus being identified as a pathogen in some cases. Before the identification of Clostridioides difficile infection (CDI), oral vancomycin was shown to effectively treat staphylococcal enterocolitis.6 In the early 1970s an outbreak of clindamycin-associated pseudomembranous colitis led to the modern era of CDI.7,8 Currently, studies estimate that there may be up to nearly half a million CDI cases every year, making it the most common bacterial cause of diarrhea in the United States.9 In spite of this number, a large proportion of AAD cases seem to be due to other causes. Studies have looked at some of the other causes that may be responsible for non-CDI AAD including other putative microbial etiologies.10,11
The likely pathogenesis of AAD includes alteration of intestinal microbiota, direct drug toxicity on the gut and development of a superinfection by a pathogenic microbe. The normal intestinal microbiota importantly contributes to intestinal homeostasis and colonization resistance by pathologic microflora by production of antimicrobial metabolites, utilization of micronutrients and space, and modulation of the gut immune system.12 Administration of antibiotics causes changes in the fecal flora and creates gaps in ecological niches that allow proliferation of opportunistic microbes. Although the role of C. difficile in the pathogenesis of AAD has been well studied, there are considerable gaps in our understanding of the pathogenesis of non-CDI AAD due to putative microbial etiologies including C. perfringens, S. aureus, K. oxytoca, and Candida species. Identifying the etiology and understanding the pathogenesis of AAD will help in improving healthcare and in reducing the economic burden.
The review will also focus on the alteration of intestinal microflora and will provide a stronger basis for the use of non-pharmacologic therapies such as synbiotics (combination of prebiotics and probiotics that benefit the host by inhibiting the growth of pathogenic bacteria and enhancing the growth of beneficial organisms) as an effective therapeutic and preventive strategy. The overall objective is to review the existing data and acquire a better understanding of the various pathophysiologic mechanisms of AAD. Specific aims are: (1) to examine the roles of various microbial organisms in etiology of AAD (2) to understand the pathogenesis of C. difficile as well as non-C. difficile AAD.
METHODS
A comprehensive literature search was undertaken in Medline via Ovid and Pubmed databases as of July 31, 2022, using the key words: Clostridioides difficile, difficile, diarrhea, antibiotic associated diarrhea, AAD, colitis, enteritis, pseudomembranous colitis, CDI, CDAD, Staphylococcus, Klebsiella, Candida and Clostridium perfringens. For each organism, studies evaluating their role in pathogenesis of AAD were evaluated. Initially, studies were eligible if they referred to diarrheal illness or enteric infection by any organism following use of antibiotics. They were then narrowed down to any aspect of pathogenesis relating to enteric infection by specific organisms such as Staphylococcus, Klebsiella, Candida, C. difficile and C. perfringens. The list was further narrowed after performing a manual search of the full text for the relevant primary studies to identify their relevance and to extract any additional data.
An example of one such search strategy employed for Candida and antibiotic associated diarrhea in PubMed was (“Candida” [MeSH terms] OR “Candida”[All fields]) AND (“antibiotic associated diarrhea” [MeSH terms] OR “AAD” [MeSH terms] OR (“antibiotic associated” [All fields] AND (“diarrhea”[All Fields] OR “diarrhoea”[All Fields] OR “colitis”[All Fields"] OR “enteritis” [All Fields] OR “enterocolitis”[All Fields])) OR “antibiotic associated diarrhea”[All fields] OR “AAD”[All Fields] OR (("anti-bacterial agents[Adverse effects] OR antibiotic-associated[All Fields] OR “AAD”[All Fields]) AND (“diarrhea”[All Fields] OR “diarrhoea”[All Fields] OR “diarrhea”[Chemically induced]).
We reviewed published original articles, case reports, case series and abstracts in peer reviewed journals. Bibliographies from review articles were also scanned. No language and date restrictions were applied. Both human as well as animal studies were initially screened. Data synthesized was used to construct evidence tables, flow charts and figures to illustrate the different pathophysiologic mechanisms involved. Additional details on salient features of studies and potential biases were mentioned in the comments section of the table. Data was evaluated for inclusion and extracted from eligible studies independently by two authors.
RESULTS & DISCUSSION
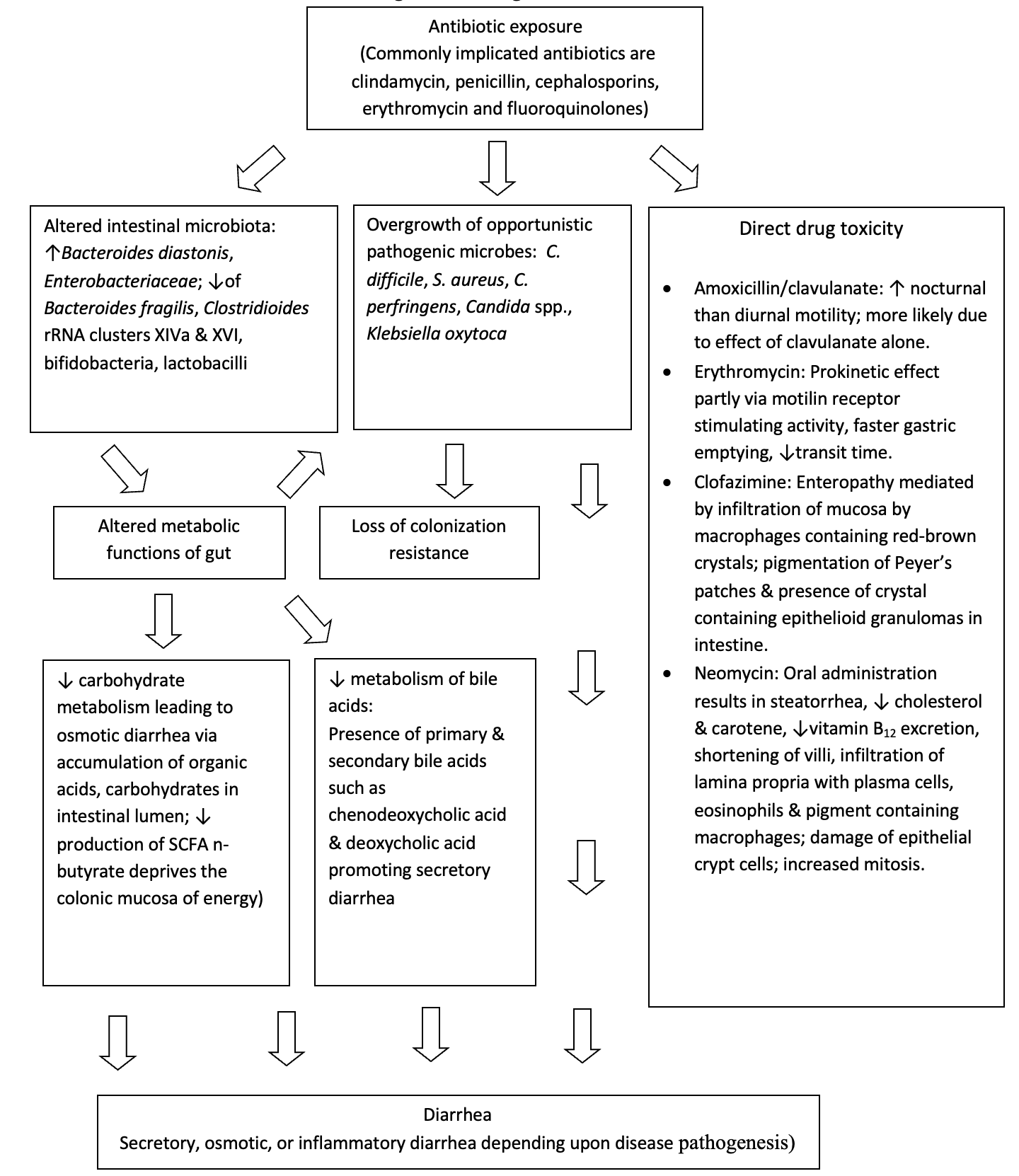
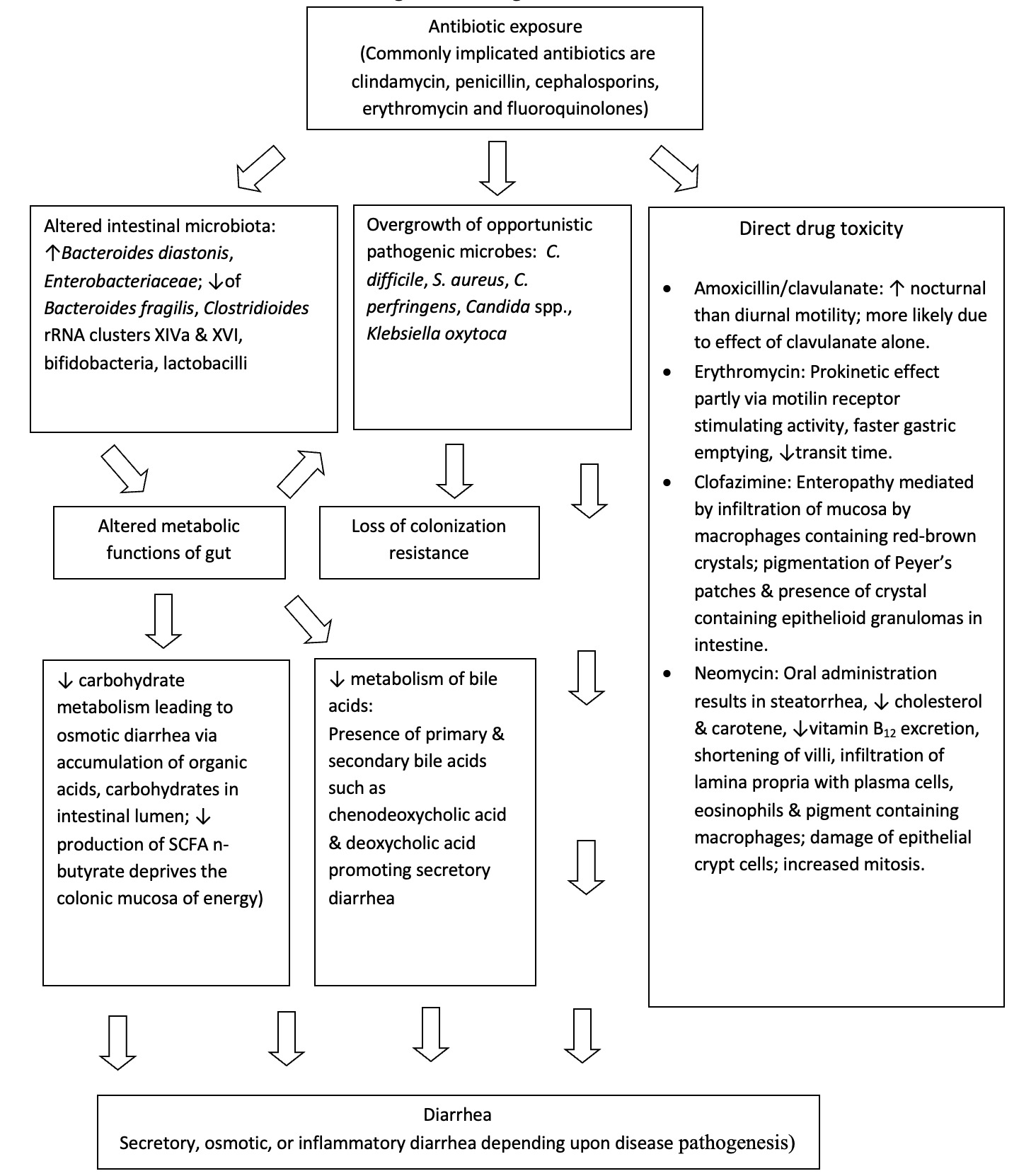
The human intestinal microbiota plays an important role in the maintenance of host health through formation of short chain fatty acids and secondary bile acids, modulation of the immune system and development of colonization resistance. Colonization resistance describes the ability of the intestinal microbiota to resist invasion by exogenous microorganisms by means of host factors such as production of inhibitory substances, attachment interference, nutrient depletion, along with the production of toxic antagonistic compounds.13 Numerous adverse effects to antibiotics have been associated with changes in the intestinal microbiota including diarrhea, acquisition of CDI, emergence of antibiotic resistant strains of enteric pathogens, and a decline in colonization resistance. Figure 1 provides an overview of the pathogenesis of AAD.
Alterations in metabolic functions of normal intestinal flora
Carbohydrates reaching the colon such as resistant starch, dietary fiber, unabsorbed sugars, and oligosaccharides undergo anaerobic fermentation in a progressive manner from proximal to distal colon. This fermentation leads to the production of short chain fatty acids (SCFAs) such as acetate, butyrate, proprionate, as well as lactic acid and gases. Altered intestinal microbiota from antibiotic exposure results in diminished absorption of carbohydrates by reducing the fermentation of SCFAs. The presence of unfermented carbohydrates overwhelms the colon mucosa and retains fluid in colonic lumen resulting in osmotic diarrhea.14 In-vitro studies have shown that even healthy subjects lose their ability to metabolize carbohydrates in the presence of antibiotics.15 Diminished production of SCFA with alteration of gut microbiota also deprives the colonic mucosa of an efficient energy source.16 Reduced concentrations of SCFA are found in healthy subjects as well as patients taking antibiotics with activity against anaerobic bacteria.17
Primary bile salts synthesized by the liver are mainly composed of cholate and chenodeoxycholate, conjugated with either taurine or glycine. The intestinal microbiota possesses bile salt hydrolases that play an important role by removing the conjugates from the primary bile salts. Deconjugated salts are dehydroxylated by 7α dehydroxylating bacteria in the large bowel to deoxycholic acid and lithocholic acid and are simultaneously precipitated from solution. Reduced amounts of 7α dehydroxylating bacteria following antibiotic exposure may result in elevated levels of chenodeoxycholic acid, resulting in secretory diarrhea.18
Microbial Causes of AAD
In Table 1, the role of putative microbial etiologies in AAD are listed. Administration of antibiotics causes alteration of intestinal microbiota with loss of colonization resistance. This results in overgrowth of organisms from exogenous origin or from other regions of the gut that may alter gut function. Antibiotic resistant endogenous organisms may overgrow with their selective advantage during antimicrobial therapy and occasionally resistant pathogens may be encouraged to grow when kept normally in small numbers by normal flora.
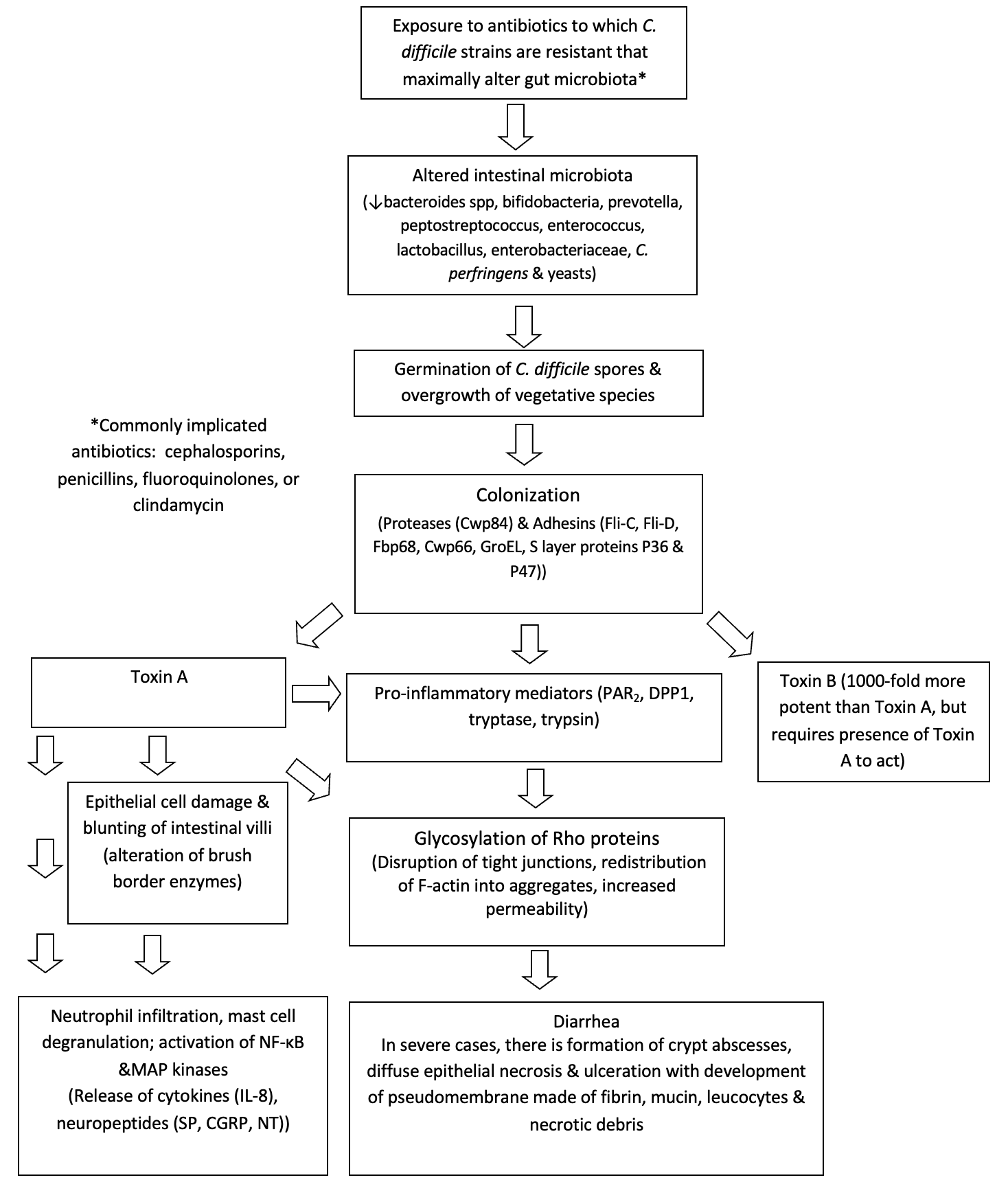
C. difficile is the most identified cause of AAD. Clinical manifestations in CDI range from short-term colonization, acute diarrhea, fulminant diarrhea, which may be associated with pseudomembranous colitis and leukocytosis and may lead to death, and recurrent CDI (rCDI).9 The severity of illness may depend on risk factors such as age, host immune status, number of prior hospitalizations and presence of co-morbidities. Major risk factors include advanced age, hospitalization, and exposure to antibiotics. Important pathophysiologic alterations seen in CDI are highlighted in Figure 2. An exposure to vegetative cells and/or spores of C. difficile, antibiotic alteration of intestinal microbiota and exposure to bile salts which convert spores to vegetative cells can lead to the propagation of C. difficile in the gut.20 Other factors involved in CDI pathogenesis include proteolytic enzymes released from the organism such as cysteine protease Cwp84 and bacterial adhesins such as flagellin Fli-C, flagellar cap protein Fli-D and Cwp 66, fibronectin binding protein Fbp 68 heat shock protein GroEL, and surface (S) layer proteins such as P36 and P47.21–29
.png)
The two main toxins of C. difficile, toxins A and B play a critical role in the pathophysiology of CDI. Both are enterotoxic and cytotoxic; however, traditionally toxin A is named “enterotoxin A” and toxin B is named “cytotoxin B”. Toxins A and B share 63% of common amino acid sequence homology and play a significant role in the mucosal inflammation and injury seen in CDI.30 Strains of C. difficile may also produce a binary toxin (or Clostridioides difficile transferase) first described by Popoff et al.31 This toxin consists of a binding component, an enzymatic component and also demonstrates actin specific ADP ribosyl transferase activity that causes cytoskeletal re-organization and cell death.32 It is frequently seen in the hypervirulent North American Phage type 1 (NAP1) (also Ribosome 027) strain of C. difficile, which was found to produce 16 times more toxin A and B than other strains. The BI/NAP1/027 strain is usually resistant to fluoroquinolones, exhibits intensive spore production, and is responsible for the most severe CDI cases.
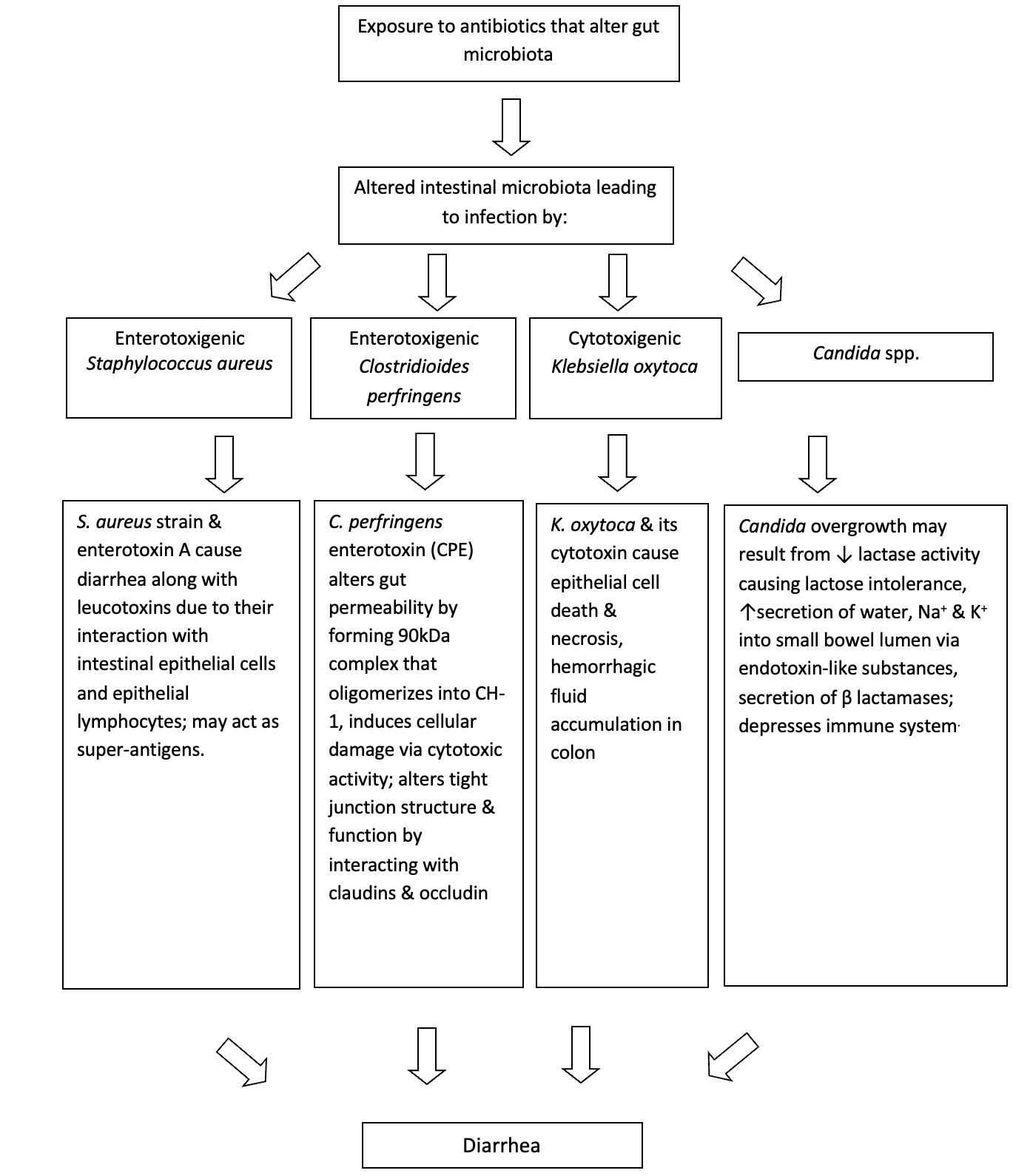
Although C. difficile is implicated in most AAD cases, other causes of Non-C. difficile Pathogen-specific AAD should also be considered (Figure 3). C. perfringens type A has also been implicated in 3-20% cases.33,34 C. perfringens AAD is primarily mediated by enterotoxins (CPEnt) released by the organism. Borriello et al. first demonstrated 11 C. difficile negative AAD cases in whom enterotoxigenic C. perfringens was detected, that were not typical of C. perfringens food poisoning.35 Ten (91%) of the patients had reported taking antibiotics three weeks before the onset of diarrhea. All stool samples had high counts (>106 cfu/g) of C. perfringens and were CPEnt positive. Clinically, these patients had more severe and protracted symptoms compared to those of food poisoning. In addition, AAD due to C. perfringens does not result in the formation of pseudomembranes. The enterotoxin gene (cpe) in most C. perfringens-associated AAD cases is primarily plasmid-borne compared to chromosomal enterotoxic gene in food poisoning cases, potentially implying that C. perfringens-AAD may be a distinct entity by itself.36,37 Prevalence studies have reported wide differences in detecting C. perfringens enterotoxin vs C. difficile toxins.19 In one study, 693 stool samples with more than 90 percent of them showing clinical evidence of AAD were prospectively investigated to determine the frequencies of C. perfringens and C. difficile.38) Interestingly, while C. difficile toxin was detected in 79 (11.4%) cases and C. perfringens enterotoxin in only 1 (0.14%) case, C. perfringens was cultured almost twice as frequently as C. difficile in this study (21.2% vs 12%). On the other hand, studies have also reported similar frequencies in detecting CPEnt and C. difficile toxins.39 In some cases, CPEnt and C. difficile toxins were found to co-exist, suggesting that they may act in synergy to cause AAD. While it appears that C. perfringens is a cause of a subset of patients with AAD, further study of the etiologic agent is needed.

S. aureus induced AAD was identified early in the antibiotic era as a cause of AAD in hospitalized patients.40 Following the identification of C. difficile as an important cause of AAD and pseudomembranous colitis, S. aureus has largely been discounted as a potential etiologic agent in AAD. However, following the widespread emergence of MRSA, investigators in Japan and Australia have reported several cases of MRSA-associated AAD.41,42 Commonly identified risk factors of Staphylococcal enterocolitis included use of antibiotics (especially fluoroquinolones), advanced age, use of H2 blockers or proton pump inhibitors, HIV infection, immune suppressive therapy and recent partial or total gastrectomy.43–49 Although pathogenetic mechanisms in S. aureus AAD have not been well appreciated, S. aureus enterotoxins (A-E & TSST-1) have shown considerable experimental evidence to cause diarrhea.50 In France, investigators reported predominant MRSA growth in stools of some suspected AAD patients, most of which produced enterotoxin A and the bicomponent leucotoxin (LukE-LukD).45 Investigators have looked for clinical differences between S. aureus induced AAD vs. CDI. In one small study patients with S. aureus- associated AAD presented with greenish watery stools not seen in patients with CDI.51 A predominance of gram-positive cocci in clusters on gram stain in stool or on mucosal biopsy along with watery diarrhea supports the diagnosis. Froberg et al. reported a patient with simultaneous infections by C. difficile and S. aureus with two distinct pseudomembranous lesions.52 With S. aureus, the pseudomembranes were loosely adherent, yellowish-green and located primarily in the small bowel compared to tightly adherent, sharply demarcated, and small, eruptive pseudomembranes located in the cecum and the colon in CDI. Investigators also reported that clinical features of patients co-infected by both pathogens are likely to be more serious than those who are infected by single pathogen.53,54 Taken together, MRSA may have a valid role in the pathogenesis of AAD, particularly in cases of small bowel involvement and/or when predominantly present in the stool. Criteria however need to be established to initiate investigations.
K. oxytoca, a gram-negative facultative aerobic enterobacterium, has been shown to be the causative agent of antibiotic associated hemorrhagic colitis (AAHC).55 Although sharing some similarities with C. difficile, AAHC appears to be a clinically distinct form of colitis that is characterized by the absence of toxigenic C. difficile and the presence of hematochezia. It is predominantly segmental, involves the right colon and is associated typically with use of the penicillins since Klebsiella expresses beta-lactamases.55,56 Clinically, patients present with bloody diarrhea during antibiotic therapy, accompanied by severe abdominal cramps and often resolve spontaneously following cessation of antibiotics.55,57 The pathogenesis is related to the production of a cytotoxin that leads to epithelial cell death and predisposes patients to hemorrhagic colitis.57 Further evidence of the pathogenesis has been seen in studies where intraluminal administration of cytotoxin from the organism resulted in direct damage to the ileal mucosa that was demonstrated histologically.58 Colonoscopy typically reveals segmental distribution of mucosal inflammation and hemorrhage, predominantly in the right colon.55
Hogenauer et al. investigated patients who had suspected C. difficile negative AAD to determine the pathogenetic role of K. oxytoca in AAHC.55 Cultures for K. oxytoca were sent following a diagnosis of AAHC on diagnostic colonoscopy. Out of 22 consecutive C. difficile-negative AAD patients, six patients had findings consistent with hemorrhagic colitis on colonoscopy. Of those, five patients had positive stool cultures for K. oxytoca. K. oxytoca was also detected in the stools of 6 of the 385 healthy subjects, who had no gastrointestinal symptoms including diarrhea. Although the role of K. oxytoca was demonstrated in AAHC, it was not known if it played a similar role in nonhemorrhagic antibiotic associated colitis. Out of 371 stool samples in patients with suspected AAD, K. oxytoca was isolated in 15 samples, and C. difficile toxin in 14 of them. Among the K. oxytoca isolates, 6 were toxin producing, of which 3 were associated with only AAHC. There were no cases of nonhemorrhagic colitis detected.
The role of Candida spp. in AAD has been debated since the 1950s and continues to be controversial. Candida spp. normally colonizes the small and large intestine in insignificant numbers, and usually does not exceed 104 cfu/ml.59 However, they are also the most frequently encountered opportunistic fungal pathogen of the gastrointestinal tract following antibacterial therapy.60 In patients treated with antibiotics, increased fecal counts of Candida have been observed and have been tentatively linked to the development of diarrhea. However, this appears to be the consequence of antibiotic therapy rather than the cause.61 Some of the risk factors commonly associated with Candida infection besides antibacterial therapy (especially tetracyclines62) are increasing age, endocrine abnormalities, immune dysfunction, chemotherapy, neoplasm, and steroid therapy.62–65 Clinically these patients present with prolonged secretory diarrhea with abdominal pain and cramping but without blood, mucus, fever, nausea, or vomiting.
Several mechanisms have been suggested to explain why Candida spp. overgrowth may be associated with AAD. Reduction in the number of endogenous intestinal microbiota may result in proliferation of Candida spp. due to lack of growth restraints and presence of selective growth advantages. One study suggested that the interruption of the colonization resistance following exposure to tetracyclines could facilitate overgrowth of undesirable organisms such as C. albicans. This was supported by no growth of C. albicans when normal intestinal microbiota was present.62 Other studies that have shown supporting evidence have suggested that the diarrhea may be due to secretory changes in the gut,63,66 acquired lactase deficiency,67 abnormal lactose metabolism68 or inhibition of leukocyte activity following antibacterial therapy.63,66–69 In vitro and animal studies have shown that Candida spp. may alter gut immune mechanisms.70,71 However, there has been no convincing evidence that supports the role of soluble Candida inhibitors such as phospholipase in the pathogenesis of Candida associated AAD.72,73 In summary, the role of Candida spp. in AAD remains controversial although there may be an association between Candida overgrowth and AAD in selected patients such as elderly inpatients who are receiving antibiotics.73–75
Only 40% of AAD can be accounted for by the above causes leaving a majority of AAD cases without an etiology. Limited studies have shown that strains of Pseudomonas aeruginosa may be a possible cause of AAD.11,76–78 Song et al. provided some evidence that strains of K. pneumonia were associated with AAD and Vaishnavi et al. identified strains of Campyobacter jejuni in 14 out of 138 (10%) patients with AAD.11,79 Strains of Salmonella and C. jejuni have been associated with an increased rate of infection in people receiving antibiotics.80–83 However, pathogenic mechanisms of organisms such as Salmonella and C. jejuni have not been well described.
Nonmicrobial Causes of AAD
Non-microbial-causes of AAD tend to occur within 48-72 hours of starting antibiotics, tend to be less severe and usually are self-limiting. Macrolides such as erythromycin can cause stimulation of motilin receptors, increase gastric emptying, and decrease transit time. Beta-lactam antibiotics can cause osmotic diarrhea due to changes in degradation of non-absorbed carbohydrates, changes in bile salt degradation, and stimulation of c-AMP which increase bowel activity through increased chloride secretion. Amoxicillin/clavulanate also may cause increased nocturnal motility. Neomycin can cause steatorrhea, reduced vitamin B12 excretion, shortening of villi, infiltration of lamina propria with plasma cells, eosinophils and pigment containing macrophages. High dose clofazimine therapy may also cause red crystal deposition in the small bowel lamina propria and may result in severe and fatal enteropathy.84–89
DIAGNOSIS
It is essential to rule out other causes of AAD such as infection. For CDI, the diagnosis is commonly made by detection of Clostridioides difficile toxins by enzyme linked immunoassay (EIA) or using DNA based tests that identify the microbial toxin genes in unformed stool. Stool cultures may be sent to rule out other pathogens, if clinically indicated. Stool culture for Clostridioides difficile requires anerobic culture and is not widely available. Posttreatment testing has no role in confirming eradication of the infection. Endoscopy is indicated if patient has typical CDI presentation with negative Clostridioides difficile test results, there is no response to standard course of antibiotics, patient has overlapping inflammatory bowel disease or when an alternative diagnosis is suspected, and direct visualization and/or biopsy of the bowel mucosa is needed.90,91
TREATMENT
The first-line treatments for AAD are discontinuation of the offending antibiotic and supportive care including maintaining adequate hydration. For non-severe CDI, oral vancomycin 125mg four times daily for 10 days or fidaxomicin 200mg twice daily for 10 days is the first line treatment. If fidaxomicin or vancomycin are not available, metronidazole 500mg three times daily for 10 days can be alternatively used. If metronidazole is used and the patient does not show improvement or there is clinical worsening, a switch to vancomycin or fidaxomicin should be considered.92
For patients with severe CDI, oral vancomycin or fidaxomicin should be used as first line treatment. In fulminant cases of CDI, oral vancomycin 500mg four times daily and intravenous metronidazole 500mg every 8 hours are recommended. For patients with complete ileus or colonic diversion, addition of rectal vancomycin should be considered. Early surgical consultation for patients with fulminant CDI should also be considered. CDI recurrence (rCDI) is defined as recurrence of symptoms and positive C. difficle test within eight weeks of completing therapy. In patients with first rCDI, patients should be treated with fidaxomicin for 10 days or prolonged vancomycin taper if vancomycin was used initially. Oral vancomycin 125mg four times daily for 10 days may be used if metronidazole was used initially. In case of second or subsequent recurrences, patients may be treated with prolonged vancomycin taper or oral vancomycin followed by rifaximin course.93
Fecal microbial therapy (FMT) should be considered in patients with two or more recurrences to correct the disruption in microbiota caused by recurrent use of antibiotics. In a systematic review conducted by Quraishi et al. in 2017, clinical resolution occurred in 92% of the patients treated with FMT.94 In another study from the Netherlands, 81% of the patients had resolution of rCDI after a single duodenal FMT infusion compared to 31% in the vancomycin group.95 Limited RCT data also exists for monoclonal antibody treatments such as bezlotoxmumab (monoclonal antibody that binds to C.difficile toxin B) where patients were noted to have lower rates of rCDI than patients treated with antibiotics alone. There are ongoing trials for use of enema based microbial replacement, capsule-based therapies and FMT using nasogastric tube in pediatric patients.96,97 Probiotic use has shown conflicting results and more research is needed.98–100
CONCLUSION
As the prevalence of AAD increases, the health and economic burden associated with this illness continues to be significant. For better treatment options to be developed and the trend of increasing prevalence to be reversed, significant gains must be made in our understanding of the etiology and pathophysiology of AAD. The unique features of the current review were to summarize the existing data on AAD and provide concise descriptions of the pathogenesis of CDI as well as non-CDI AAD. Because a majority of the AAD cases are due to altered intestinal microbiota, modulating the microbiota, and adopting preventive strategies such as diet, prebiotics, probiotics, and drugs may be ultimately necessary to ensure optimal health and favorable outcomes in disease prone individuals. Future research must be directed towards identifying and better understanding some of the mechanisms that operate behind benign causes as well as other microbial causes of AAD. Efforts must also be made towards establishing criteria to identify at risk patients and to initiate investigations in these patients to ensure optimal utilization of resources and to reduce the economic impact.
Author Contribution
All Authors (VS, MAA) have reviewed the final manuscript prior to submission. All the authors have contributed significantly to the manuscript, per the ICJME criteria of authorship.
-
Substantial contributions to the conception or design of the work; or the acquisition, analysis, or interpretation of data for the work; AND
-
Drafting the work or revising it critically for important intellectual content; AND
-
Final approval of the version to be published; AND
-
Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Disclosures/Conflicts of Interest
The authors have no conflicts of interest to disclose.
Funding Statement
No funding was obtained for this manuscript.
Corresponding Author
Vijairam Selvaraj MD, MPH
164 Summit Ave
Department of Medicine
The Miriam Hospital and Warren Alpert Medical School of Brown University
Providence, RI 02906
Email: vijairam.selvaraj@lifespan.org
Ph: 401-793-2500
Fax: 401-793-4047
ORCID: 0000-0002-8507-9891