BACKGROUND
Symptoms of amyloidosis may be confused in the elderly with those found in more common medical disorders. The average delay in diagnosis from symptom onset is seven to eight months. Lack of recognition of amyloidosis, especially the varied multi-systemic symptoms, contributes to this delay. We present a unique case of an elderly male who presented with an eight-month history of progressive lower extremity edema and dyspnea and was ultimately diagnosed with AL amyloidosis and concurrent renal cell carcinoma (RCC).
CASE PRESENTATION
A 73-year-old male with a history of hypertension presented following a syncopal event. This was preceded by an eight-month history of slowly progressive anasarca, severe resting and exertional dyspnea, and fatigue. He also reported gaining approximately 20 pounds over three months. His past medical history was significant for hypertension and a remote twenty-pack year smoking history. His home medications included amlodipine 10mg daily and furosemide 20 mg daily.
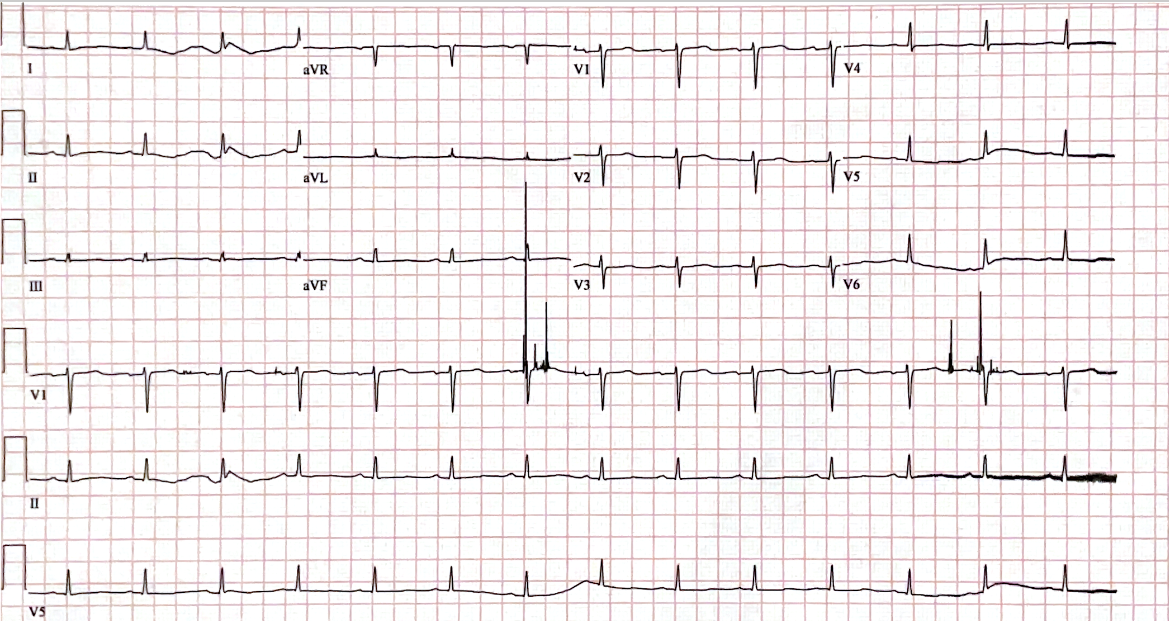
On admission, his blood pressure was 100/60, his heart rate was 120 beats/min, and his oxygen saturation was 92% on ambient air. On examination, there was dullness to percussion and diminished breath sounds at the lung bases. The examination of the lower extremities was significant for 4+ pitting edema, and the patient also had widespread swelling of the soft tissue of his trunk, scrotum, and periorbital regions. Laboratory testing revealed acute renal failure (serum creatinine was 2.1 mg/dl, and urine testing revealed nephrotic range proteinuria, with a urine protein level of 206 mg/dL (normal 0-13.9 mg/dL), and urine protein/creatinine ratio of 7.2. Serology revealed normal viral hepatitis serology, human deficiency virus, antineutrophil cytoplasmic antibodies, and complement levels. His brain natriuretic peptide was 950 pg/mL (normal <125 pg/ml), albumin was 1.7 g/dL (3.2-4.8 g/dL), the thyroid-stimulating hormone was 38.15 uIU/ml (0.35-5.5 uIU/ml), and the free T4 level was 0.64 ng/dL (0.89-1.76 ng/dL). An electrocardiogram demonstrated low voltages (Figure 1). A chest radiograph demonstrated moderate pulmonary edema and bilateral pleural effusions. An echocardiogram revealed biventricular wall thickening, biatrial enlargement, and an ejection fraction of 30% (all new findings compared to the normal echocardiogram six months prior). Serum immunofixation revealed abnormalities in kappa light chain 40.95 mg/L (normal 3.3-19.0 mg/L) and lambda light chain free 118.2 mg/L (normal 5.71-26.3). No monoclonal proteins were detected.

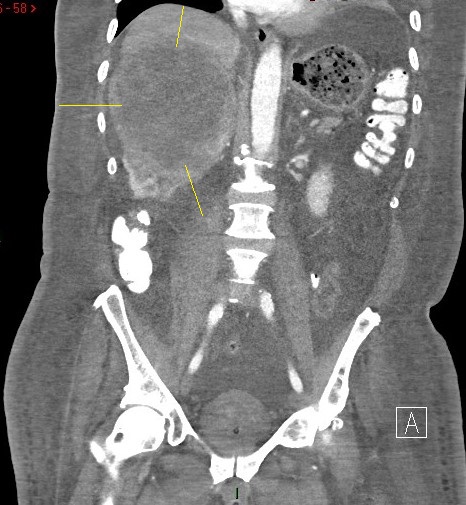
A computed tomography scan of the chest/abdomen/pelvis revealed a 13 x11.2 cm mass in the middle upper pole of the right kidney with moderated diffuse anasarca (Figure 2). A renal resection was performed, and histologic examination confirmed AL amyloidosis (positive congo red staining with involvement of glomeruli) coupled with incidental papillary stage 1 RCC. The immunofluorescence studies demonstrated staining with IgM, C3, and lambda with negative kappa. The mass spectrometry confirmed AL amyloidosis. Finally, a bone marrow biopsy was done that excluded lymphoproliferative disorders, MM, infection, and hemochromatosis.

Aggressive diuresis with furosemide was initiated for pulmonary edema. Levothyroxine was initiated for profound hypothyroidism. For the amyloidosis, the patient was treated with CyBorD (cyclophosphamide + bortezomib + dexamethasone). The underlying RCC was determined to have low aggressiveness, so therapy was focused on the cardiac and renal amyloidosis for four months. Unfortunately, he did not have favorable hematologic or clinical responses, and his functional status continuously declined, with a Karnofsky score of 30 at the conclusion of therapy. A follow-up echocardiogram revealed worsening cardiomyopathy (EF 15-20%). Cardiology and oncology specialists recommended transitioning to comfort-focused care. Unfortunately, the patient died seven months after his initial diagnosis.
DISCUSSION
Amyloidosis is characterized by extracellular deposition of abnormally folded proteins in diverse organs (most commonly the heart and kidneys).1–5 Mature fibrils possess binding sites for indicator dyes - Congo red, which is demonstrable under polarized light.2,3 Virchow first coined the term amyloid in 1854. As a disorder of protein metabolism, amyloidosis is rare, heterogenous, and poorly understood. Prasad, et al. reported a nearly identical case in a 77-year-old female who presented with congestive heart failure, nephrotic syndrome, AL amyloidosis and RCC.1
AL amyloidosis, the most common type of systemic amyloidosis in the developed world, is an underlying plasma cell disorder characterized by excessive secretion of immunoglobulin light chains that deposit into affected organs.1–4 Approximately 20% of patients with AL amyloidosis present with coexisting multiple myeloma (MM), while the rest are diagnosed with plasma cell dyscrasias.6 The diagnosis is often difficult because AL amyloidosis may present with near-normal blood counts and polyclonal plasma cells.3,6 MM is closely associated with AL amyloidosis because in each condition, identical clones of antibody producing cells multiply, and the treatments are similar. AL amyloidosis occurs more commonly in the elderly. The average age of a patient with AL amyloidosis is 64 years, with an age range of (50-80 years), although there are some case reports of persons in their twenties who were diagnosed with AL amyloidosis.7 Two-thirds of the elderly patients with AL amyloidosis are male.
The clinical diagnosis of AL amyloidosis is quite challenging and often delayed due to variable, nonspecific symptoms depending upon which organs are involved. A high index of clinical suspicion is required to diagnose amyloidosis.6,8 Affected organs include the heart (70%), kidney (60%), peripheral and autonomic nervous system (15%), gastrointestinal (15%), liver (20%) and soft tissue (10%).1–3,6 Clinical presentations that should immediately elicit consideration of amyloidosis includes non-diabetic nephrotic range proteinuria, cardiac failure without aortic stenosis or hypertension, or unexplained peripheral neuropathy. Very specific signs such as macroglossia and periorbital purpura is often not present.6 The median time from symptoms onset to clinical diagnosis is approximately seven months.6 Earlier amyloidosis diagnosis correlates with improved clinical outcomes because of end organ damage.1–4,6
The degree of cardiac involvement is the most reliable predictor of survival in AL amyloidosis. The most common electrocardiogram change seen with amyloidosis is low voltages in the limb leads. Li, et al. reported that abnormalities in electrocardiogram parameters may provide potential prognostic value in patients with renal AL amyloidosis, that may occur with infiltration and restriction of the heart from cardiac amyloidosis.7 Specifically, prolonged PR and QT intervals on electrocardiogram are associated with an increased all-cause mortality in renal AL amyloidosis, which correlate to results seen on echocardiogram or endomyocardial biopsy.7
RCC accounts for 3% of adult malignancies and is often referred to in the medical literature as "the great masquerader.9,10 This is because RCC is frequently associated with multi-systemic paraneoplastic syndromes (10-40% of patients). The triad of (hematuria, flank pain, and palpable abdominal mass) is discovered in only 10-15% of patients at presentation and, when present, usually signifies advanced disease.9,10 RCC remains clinically silent for long time periods due to the kidney’s retroperitoneal location. Because of tumor production of interleukin 6 (IL-6) and TNF alpha tumor, constitutional symptoms such as fever, asthenia, cachexia, and weight loss may arise.9,10 RCC is frequently a disease of the elderly, and the incidence rises sharply with age.9
IL-6 produced by RCC is a potential para-neoplastic inflammatory link to MM.9–11 In a case series of 119 patients with metastatic RCC, IL-6 levels were significantly higher in those with paraneoplastic fever and weight loss. High levels of IL-6 in RCC patients are correlated with worse survival and drug resistance.9,10 Interestingly, IL-6 is also implicated in MM as a pro-inflammatory growth factor crucial in the proliferation and survival of MM cells and plasma cell dyscracias.11 Thus, we believe our patient’s simultaneous diagnoses of RCC, and amyloidosis were more than a coincidence. Blockade of IL-6 has been proposed for therapy for MM and RCC. Furthermore, resection of the malignancy may ameliorate or even potentially reverse the biochemical processes which involve IL-6 that drive the amyloidosis.9–11
CyBorD has been the standard of care for managing AL amyloidosis, which has shown high rates of hematologic response and prolonged progression-free survival when used in newly diagnosed patients and at relapse.12,13 Novel plasma cell-directed therapies for AL amyloidosis are emerging due to observed success in MM, which has been show to achieve hematologic control and suppress pathologic light chain precursor protein production.12–14 Daratumumab, combined with CyBorD (CyBorD-DARA), is now the first US Food and Drug Administration (FDA) approved treatment for AL amyloidosis for patients newly diagnosed with AL amyloidosis. Approval was based on the Phase 3 (ANDROMEDA) trial.15 Unfortunately, poor treatment outcomes in elderly may be due to their advanced ages, time delay in diagnosis, aggressive pathogenesis of AL amyloidosis, and poor Karnofsky performance scores.
In summary, a high level of suspicion for amyloidosis is important due to its variable nonspecific symptoms. In patients with established malignancy especially RCC, amyloidosis should be on the differential diagnosis of unexplained congestive heart failure or unexplained nephrotic syndrome. Amyloidosis should be included as a potential paraneoplastic manifestation of RCC because earlier therapy may result in improved outcomes.
ACKNOWLEDGEMENTS
The authors would like to acknowledge Karolina Lickunas for the editorial review and drafting of portions of the manuscript.
DISCLOSURES/CONFLICTS OF INTEREST
The authors have no conflicts of interest to disclose.
AUTHOR CONTRIBUTION
All Authors have reviewed the final manuscript prior to submission. All the authors have contributed significantly to the manuscript, per the ICJME criteria of authorship.
-
Substantial contributions to the conception or design of the work; or the acquisition, analysis, or interpretation of data for the work; AND
-
Drafting the work or revising it critically for important intellectual content; AND
-
Final approval of the version to be published; AND
-
Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved
CORRESPONDING AUTHOR
Hien Nguyen, MD
6104 Old Branch Avenue
Temple Hills, MD, 20746
Telephone 301-702-6100
Fax 301-702-6367
Email: Hien.X.Nguyen@kp.org