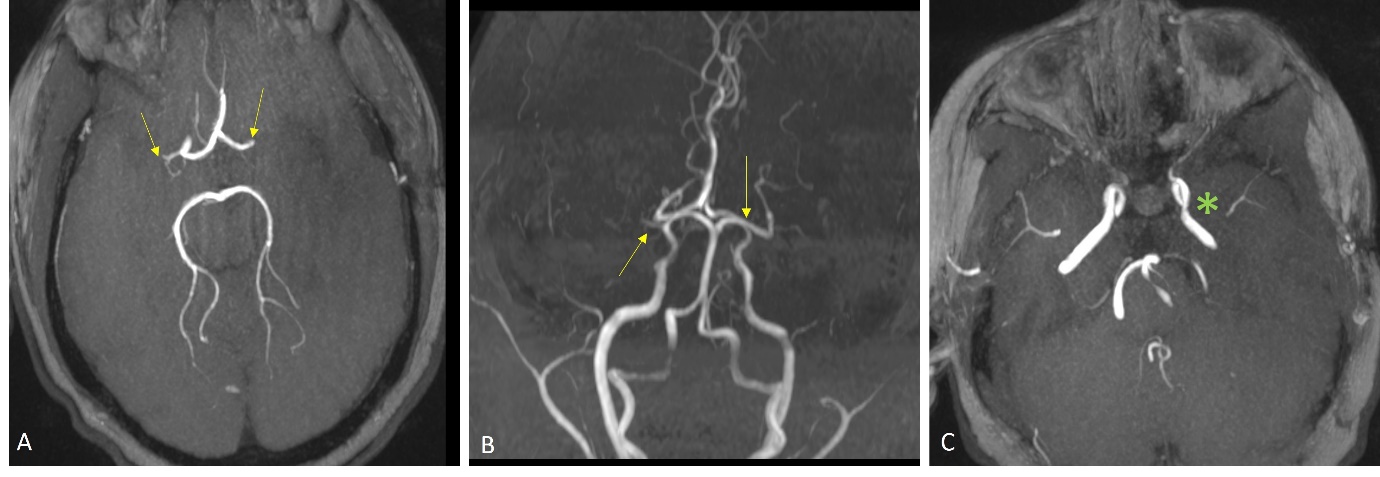
A 13-year-old girl presented with sudden onset of expressive aphasia and right hemiplegia. Her past medical history included type 1 diabetes mellitus on insulin pump therapy and hypertension treated with lisinopril. Her mother had insulin-dependent diabetes mellitus. Her National Institutes of Health Stroke Scale on arrival was 23/30. Computed tomography of the head showed a hypodensity area in the left parietal lobe. The onset of symptoms was outside the time window for intravenous thrombolysis. Magnetic resonance imaging (MRI) imaging revealed subacute infarct in the left middle cerebral artery (MCA) distribution and bilateral chronic lacunar infarcts. Magnetic resonance angiography (MRA) demonstrated bilateral MCA M1 occlusions (Figure 1), suggestive of Moyamoya disease.
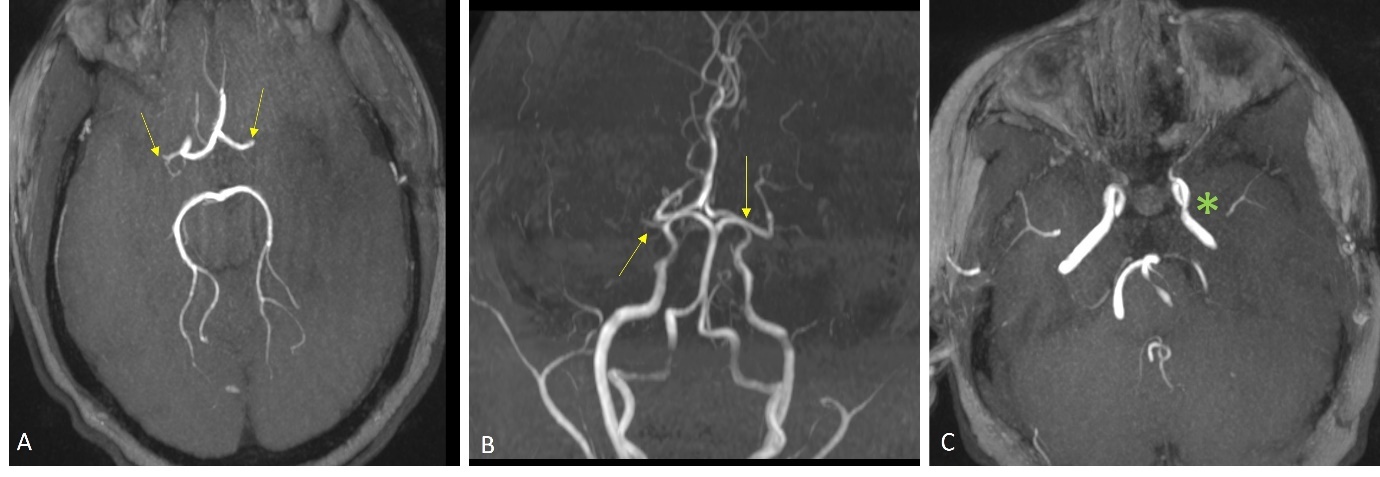
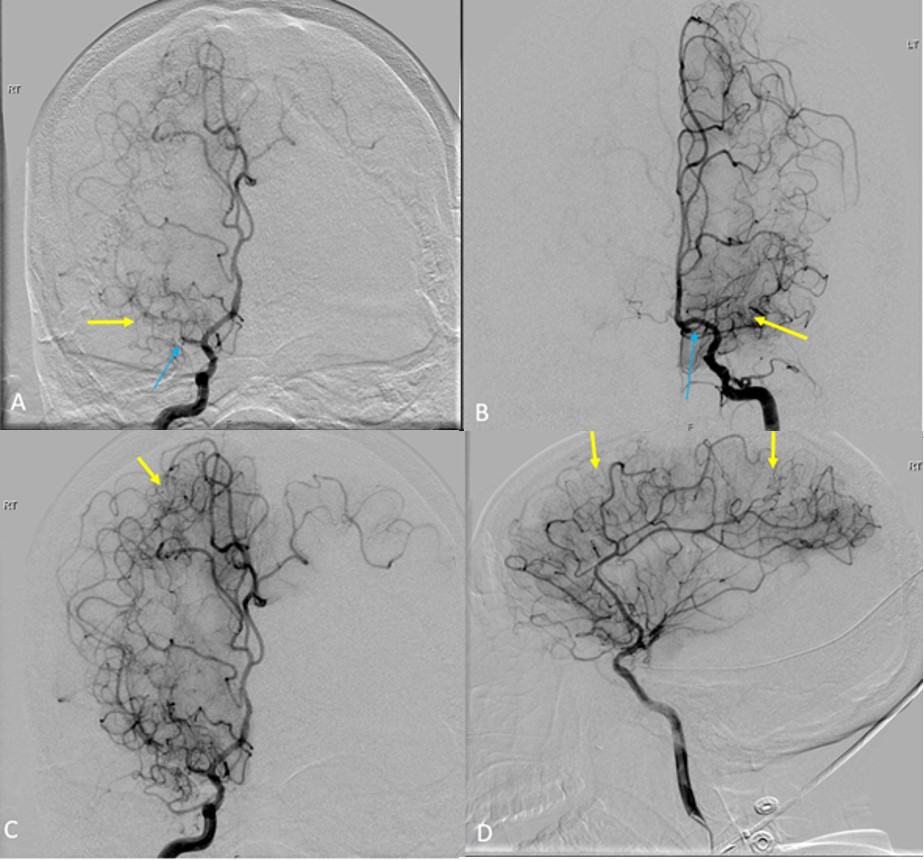
There were no collateral vessels and the bilateral supra-clinoid internal carotid arteries (ICAs) were relatively spared. Perfusion imaging showed a mismatched diffusion-positive defect, and a normal perfusion pattern for the occluded right M1 segment of MCA. MRI imaging of the vessel wall was performed. It showed minimal enhancement of the proximal left M1 segment and sluggish blood flow in the distal MCA, findings which favored Moyamoya disease and pointed against intracranial atherosclerotic disease. She underwent catheter angiography- which showed complete occlusion of the right M1 segment at the ICA termination and robust leptomeningeal collateral derived from the right anterior cerebral artery. (Figure 2)

4 months later, patient underwent craniotomy for a left encephaloduroarteriosynangiosis (EDAS) for collateralization of the left anterior circulation. She was lost to follow-up and presented three years later with worsening kidney function. MRI brain showed progression of the internal carotid arteries stenoses without new neurologic deficits. The patient expired due to acute hypoxic respiratory failure secondary to pneumonia.
Moyamoya disease is a rare cerebrovascular disease characterized by progressive stenosis of the distal bilateral internal carotid arteries and their proximal branches with compensatory lenticulostriate collaterals that is described as “a puff of smoke” (Moyamoya in Japanese).1 Moyamoya syndrome is the association of Moyamoya-like vasculopathy with systemic diseases including neurofibromatosis type 1, Down’s syndrome, cranial irradiation, sickle cell disease, and hyperthyroidism.2 Moyamoya disease has a bimodal age distribution: the first peak in the 1st decade and a second peak in the 4th – 5th decade, with a 2.9:1 female preponderance.3 Children commonly present with ischemic stroke whereas adults usually present with haemorrhagic stroke.1 Clinical manifestations of Moyamoya disease include transient ischemic attack, cerebrovascular accident, seizure, headache, choreiform movements, cognitive dysfunction and psychiatric changes.2 Genetic predisposition is implicated in the pathogenesis of Moyamoya disease, and individuals with first-degree family history of Moyamoya disease has 132-fold higher risk of developing the disease than those without.4 There is no curative treatment for Moyamoya disease. Symptomatic, supportive treatment such as maintaining euvolemia to maintain sufficient cerebral blood flow is essential. Aspirin has been used to prevent microemboli in ischemic Moyamoya disease, but it is not recommended for asymptomatic Moyamoya disease. For symptomatic patients, bypass surgery is offered to increase cerebral blood flow reserve and reduce the incidence of stroke.5
Corresponding Author
Jia Wei Tan
457 Mill Hill Ave
Bridgeport, 06610 CT
4753452692
Jiawei.tan@bpthosp.org