INTRODUCTION
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is an autosomal dominant disease which causes early onset stroke and dementia and is associated with diffuse white-matter abnormalities on neuroimaging.1 It is caused by NOTCH3 mutation on chromosome 19; hypertension, hyperlipidemia, and smoking are additional risk factors for its developement.2 CADASIL has four cardinal features including recurrent ischemic events, migraines with aura, mood disturbances and progressive vascular dementia.3 Brain magnetic resonance imaging (MRI) displays extensive white matter hyperintensities (WMH) in the anterior temporal lobe and external capsule.2 Global prevalence is 2 to 5 out of every 100,000 persons. CADASIL is underdiagnosed or often misdiagnosed as multiple sclerosis.4 Variation in genotypes has been associated with different phenotypes and ethnicities; the prevalence rate of CADASIL in Japan is estimated 1 to 3 per 100,000 adults.5 Currently there is a lack of published data from the South Asian region to determine the prevalence of CADASIL. We report a 66-year-old woman with genetically proven CADASIL and multiple co-morbid conditions.
CASE REPORT
This 66-year-old female with past history of diabetes mellitus type 2 presented to the neurology clinic in November 2021, with complaints of vertigo on bending forward for the last 6-7 years, off and on dizziness and imbalance, recurrent migraines involving the right half of the face and periorbital region for the past year, and occasional altered taste. She had three transient ischemic attacks in one week five years back, affecting the left side of her body causing weakness and mild slurring of her speech. She complained of subjective memory problems and anxiety. Her father died 5 years prior at age of 69, with a history of myocardial infarction one year prior to death but detailed examination and genetic testing were not conducted.
On physical examination, she was slow to follow instructions and made poor eye contact. Muscle power was Medical Research Council (MRC) grade 5/5 in all 4 limbs. Cranial nerve, cerebellar, sensory and postural examination was unremarkable. The relevant laboratory workup is shown in Table 1. A previously performed MRI brain with contrast in April 2021 revealed bilateral confluent symmetrical white matter hyperintensities in frontoparietal, occipital and temporal lobes; abnormal signal in external capsule and posterior limb of internal capsule; and acute lacunar infarct in left parietal region. Further workup was indicated to rule out structural brain abnormalities, stroke risk factors, depression and CADASIL. She was prescribed rosuvastatin, dual antiplatelet therapy, escitalopram for anxiety and metformin. Genetic testing for CADASIL was recommended.
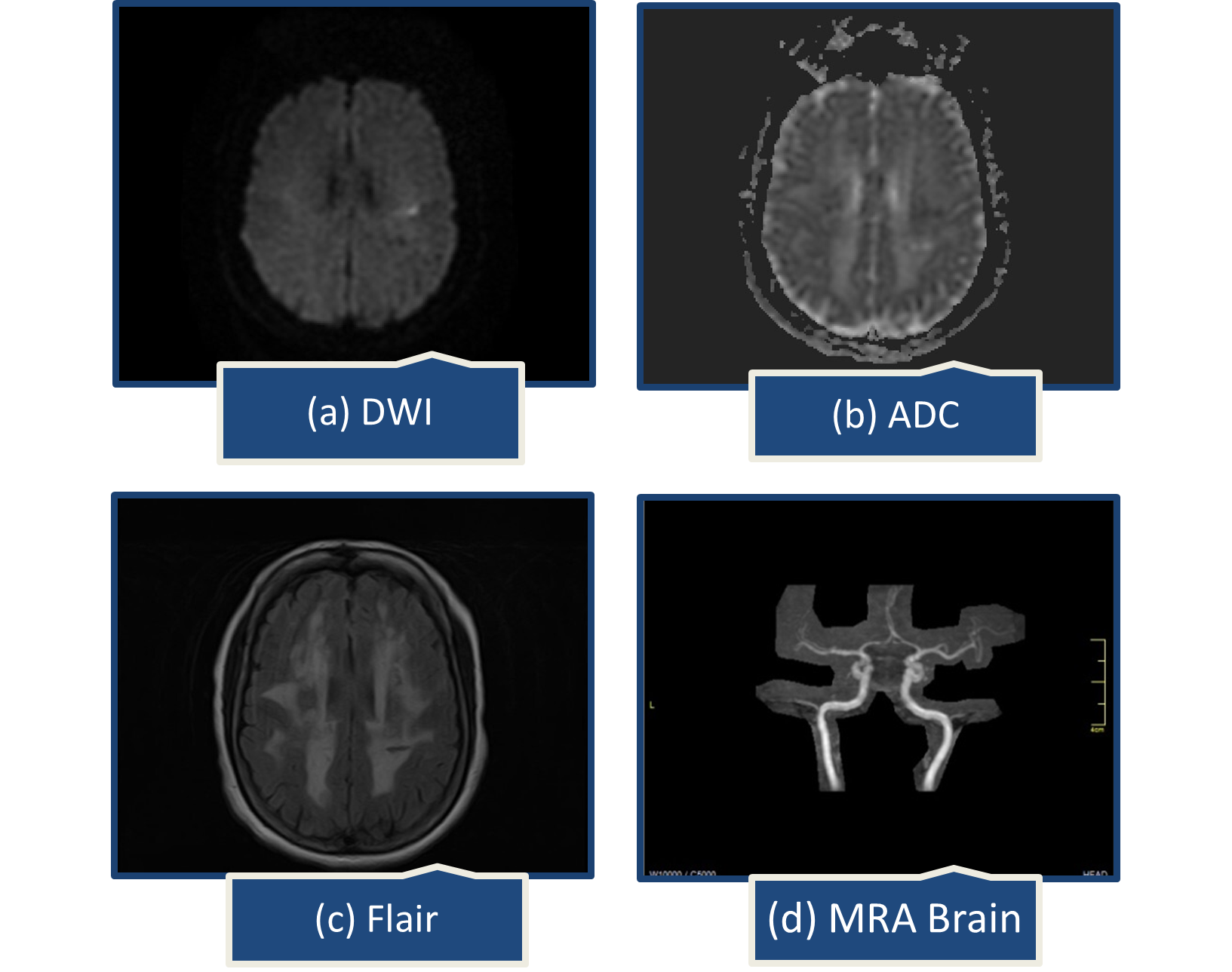
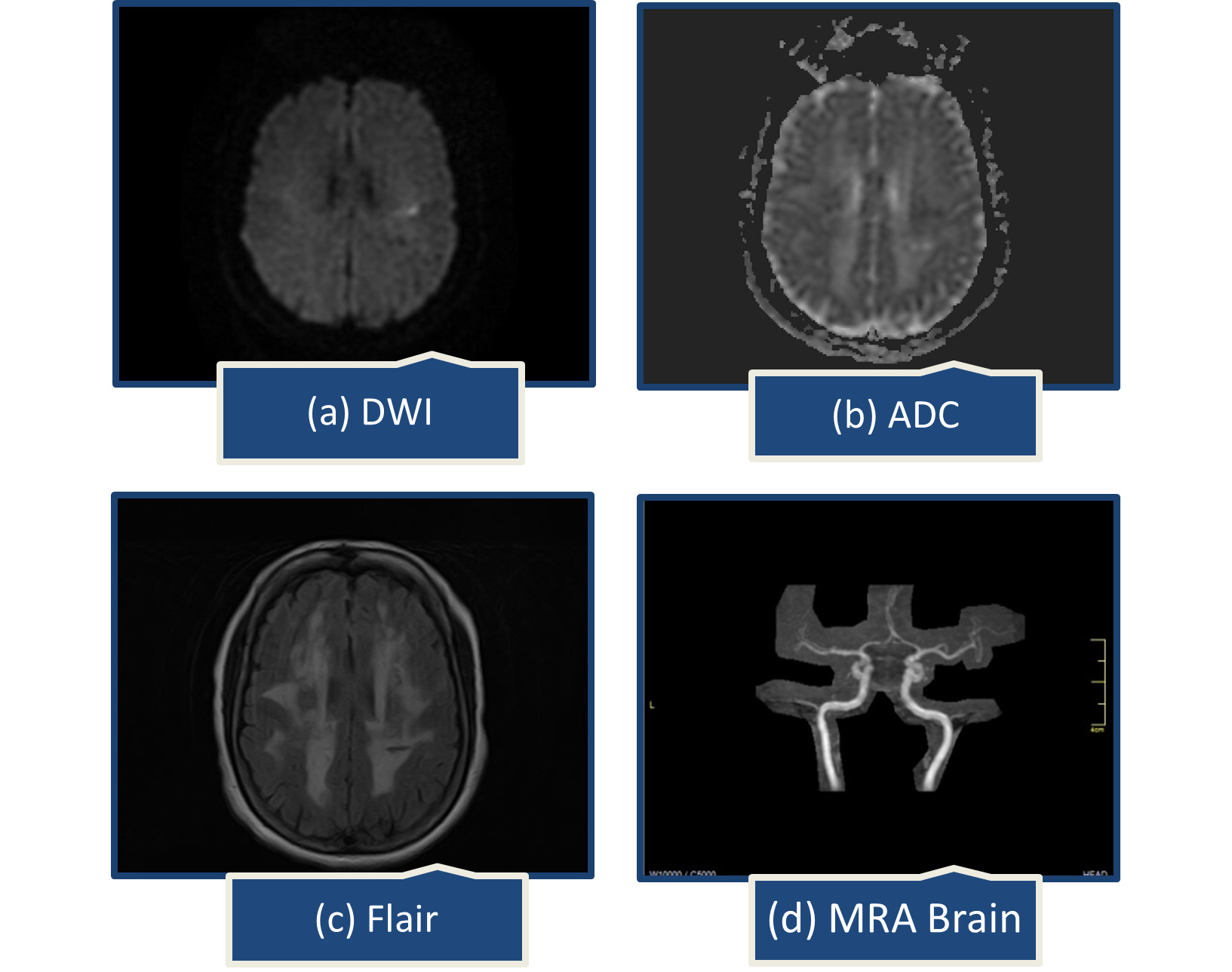
A year later, she presented with abrupt speech problems and ataxia. She could speak, but had trouble finding words, which had spontaneously improved by the time she arrived at the emergency room. There were no complaints of body weakness, numbness, or facial deviation. On examination, her Glasgow coma scale was 15/15, all reflexes intact, muscle power was MRC grade 5/5 in all limbs, National Institutes of Health Stroke Scale (NIHSS) was 2, one point for language causing mild-to-moderate aphasia; some obvious loss of fluency, without significant limitation on form of expression and one point for dysarthria causing mild-to-moderate dysarthria; patient slurred at least some words which could be understood with some difficulty. Brain computed tomography (CT) scan showed extensive confluent hypodensities in bilateral periventricular white matter. On brain MRI, a small acute ischemic infarct in left centrum semiovale and extensive confluent fluid attenuated inversion recovery (FLAIR) bright signal foci were noted in bilateral periventricular and subcortical white matter representing chronic microvascular ischemic changes (Figure 1).
She was discharged the next day with instructions to take sitagliptin/metformin 50/500 mg twice daily, aspirin/clopidogrel 75/75 mg, rosuvastatin 20 mg, and escitalopram 10 mg.
On a one-week follow-up visit, she was alert and oriented in time, place, and person. Her speech and gait were also normal. Deoxyribonucleic acid (DNA) from peripheral blood of the patient was subjected to Hereditary Cerebral Small Vessel Disease Panel (Invitae, USA). Sequence analysis and deletion/duplication testing of the 10 genes of this panel revealed a heterozygous variant, c.1136G>C (p.Cys379Ser), in exon 7 of NOTCH3 gene. This G to C missense mutation at cDNA nucleotide 1136 replaced cysteine with serine at position 379 in the NOTCH3 protein. As per American College of Medical Genetics and Genomics (ACMG) guidelines, this mutation is considered “Likely Pathogenic.” Genetic counselling for her children was performed. Both of our patient’s children were offered free of cost genetic testing, her son opted for it and tested negative.
DISCUSSION
CADASIL is characterized by more than 280 missense mutations in exons 2-24 of the NOTCH3 gene on chromosome 19p13, largely affecting the 34 epidermal growth factor receptor (EGFr) domains, which are expressed in vascular smooth muscle and pericyte cells in adults.1,3 NOTCH3 gene mutations result in severe cerebral vasculopathy, impacting arterial vascular smooth muscle cells as well as pericyte-endothelial interactions.6
Multiple small, deep cerebral infarcts are discovered during pathological analysis, along with leukoencephalopathy and a non-atherosclerotic, non-amyloid angiopathy that primarily affects small cerebral arteries.1 Ultrastructural analysis demonstrates severe alterations in vascular smooth-muscle cells.1 While elastin and amyloid staining are negative, periodic acid-Schiff (PAS) staining reveals the presence of glycoproteins within vessel walls in CADASIL.1
Progressive vascular lesions in CADASIL are characterised by the loss of anchorages and fragmentation of pericytes and smooth muscle cells, and thickening of the vessel wall without a decrease in lumen.6 Damage or lack of arteriolar and capillary pericytes is critical in the pathophysiology of the disease. Pericyte-mediated cerebral venous insufficiency explains white matter lesions and increased perivascular spaces. Pericyte disintegration and capillary damage lead to vessel wall changes, lacunar infarctions, and microbleeds found in CADASIL.6
Exons 3, 4, 5, and 8 account for two-thirds of all documented mutations, whereas exons 2, 6, 11, and 18 account for the remaining one-third.7 According to the global regions, the reported distribution of mutations is changing. Exon 11 and exon 4 NOTCH3 mutations were more frequently seen in Asian populations.7 Haemorrhagic strokes (HS) and cerebral micro bleeds (CMBs) are more common among Asians. CADASIL has less frequent expression of migraines, seizures, mental problems, and dementia in Asian individuals.7 Asians are more likely to experience TIA or ischemia symptoms, as well as cognitive impairment. However, migraine and psychiatric instability is more common in Caucasians.7
For the diagnosis of CADASIL, MRI WMH in the anterior temporal pole and external capsule have a sensitivity and specificity of approximately 90%, which lead to clinical suspicion of CADASIL.2
CADASIL mutation in NOTCH3 exon 7 is uncommon around the world; however, several cases have been documented in Asia. A C388Y mutation in the exon 7 of Notch3 was discovered in a Japanese patient who initially had upper airway viral infection and a mild headache.8 Several reports from India showed similarities to the typical symptoms of CADASIL, including headaches, deteriorating cognitive function, transient ischemic attacks (TIAs), and strokes.9,10 Only one other case of CADASIL has been reported from Pakistan, presenting with migraines, which is a common symptom of this disease.11 Our patient’s initial symptoms were vertigo, imbalance, and dizziness, which progressed to headaches, relapsing transient ischemic attacks (TIAs), subjective memory loss, and anxiety. This is a rare and underreported initial manifestation of this disease. Vertigo is an uncommon symptom of CADASIL, occurring in less than 7% of cases, and can mimic Meniere’s disease.12 Vertigo may affect up to 25% of Chinese mainland patients.12 Nearly two-thirds of CADASIL patients eventually experience relapsing TIAs.3
Mutations in NOTCH3 are reported to cause a variety of phenotypes including CADASIL-1, lateral meningocele syndrome and myofibromatosis infantile.13 This c.1136G>C (p.Cys379Ser) mutation in exon 7 of the NOTCH3 gene is a rare heterozygous form of CADASIL. Only one other instance has recently been reported in a person of Chinese ethnicity with gait instability and progressive dizziness, exhibiting the same symptoms similar to our case, except for vertigo.14 Based on advanced modelling of protein sequence and biophysical property, this missense mutation is likely to affect NOTCH3 protein function.
CADASIL currently has no known cure. Anti-platelet agents, statins and anti-depressants are used as disease modifying agents.2 While some CADASIL patients live into their seventies, male gender is associated with early immobilisation and mortality.15 In suspected cases, genetic testing can help confirm the diagnosis.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding Statement
No funding was obtained for this manuscript.
Author Contributions
All authors have reviewed the final manuscript prior to submission. All the authors have contributed significantly to the manuscript, per the International Committee of Medical Journal Editors criteria of authorship.
-
Substantial contributions to the conception or design of the work; or the acquisition, analysis, or interpretation of data for the work; AND
-
Drafting the work or revising it critically for important intellectual content; AND
-
Final approval of the version to be published; AND
-
Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Acknowledgements
We would like to thank the patient and her family for their cooperation and consent to participate in this report.
IRB#075-23, approved by our Institutional Review Board and Ethical Committee for publication of this case report.
Corresponding Author
Professor Arawakan Ahmad MD, Neurology
Shifa International Hospital, Shifa Tameer-e-Millat University, H-8/4, Islamabad, Pakistan.
Email: arsalanahmad65@gmail.com